

Genome-Wide Linkage and Association Scans for Quantitative Trait Loci of Serum Lactate Dehydrogenase—The Framingham Heart Study
- Jing-Ping Lin linj{at}nhlbi.nih.gov1
- Gang Zheng zhengg{at}nhlbi.nih.gov1
- Jungnam Joo jooj{at}nhlbi.nih.gov1
- L. Adrienne Cupples adrienne{at}bu.edu2
Abstract
Serum lactate dehydrogenase (LDH) is used in diagnosing many diseases and is significantly determined by genetic factors.
Three genes coding for LDH isoenzymes were mapped to chromosome 11q15 and 12p12. We used 330 Framingham Heart Study largest
families for microsatellite linkage scan and 100K SNPs association scan to determine quantitative trait loci of LDH level.
We estimated the heritability at 41%. Our genome-wide linkage analysis yielded several chromosomal regions, other than 11q
and 12p, with LOD scores between 1 and 2.5. None of the 100K SNPs with a P-value 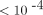 in our genome-wide association study was close to the chromosomal regions where the LDH genes reside. Our study demonstrated
a strong genetic effect on the variation of LDH levels. There may not be a single gene with a large effect, instead may be
several genes with small effects in controlling the variation of serum LDH. Those genes may be located on chromosomal regions
that differ from where the genes encoding LDH isoenzymes reside.
in our genome-wide association study was close to the chromosomal regions where the LDH genes reside. Our study demonstrated
a strong genetic effect on the variation of LDH levels. There may not be a single gene with a large effect, instead may be
several genes with small effects in controlling the variation of serum LDH. Those genes may be located on chromosomal regions
that differ from where the genes encoding LDH isoenzymes reside.
1. Introduction
LDH is a cytoplasmatic enzyme found in almost all body tissues. A small amount of the enzyme exists in the blood. When cells are damaged or destroyed, they release LDH into the bloodstream and cause blood levels to rise. LDH is used in diagnosing heart, liver, muscle, lung diseases, and testicular cancer. Its sensitivity and specificity in helping to diagnose acute myocardial infarction are very high [1].
The enzyme is composed of 4 peptide chains of 2 types, A and B. A is encoded by LDH A gene and predominant in muscle, and B by LDH B gene predominant in the heart. Five different combinations of A and B compose five LDH isoenzymes: LDH1 (A4), LDH2 (A3B), LDH3 (A2B2), LDH4 (AB3), and LDH5 (B4). While all of the five isoenzymes are represented in total serum LDH; LDH2 makes up the greatest percentage. In addition, there is homotetrameric LDH C isoenzyme, encoded by the LDH C gene, found only in mature testes and spermatozoa in humans. The LDH A and C genes are syntenic on human chromosome 11q15 [2, 3] while the LDH B gene maps to chromosome 12p12 [4].
Family studies have shown that serum LDH variation is significantly determined by genetic factors with heritability estimate as 50% [5]. So far, no genome-wide linkage or association studies on serum levels of LDH have been reported. We carried out a 10 cm genome-wide linkage analysis and an Affymetrix Human GeneChip 100 K genome-wide association study for quantitative trait loci of serum LDH level in a community-based Caucasian cohort, the Framingham Heart Study.
2. Subjects and Methods
The Framingham Heart Study, a population-based study, began in 1948 with the recruitment of 5209 residents aged 28–62 years (mean age 44.1) from Framingham, Massachusetts [6]. The participants have undergone biennial examinations since the study began. In 1971, the Framingham Offspring Study [7] was started, in part, to evaluate the genetic components of cardiovascular disease etiology. In total, 5124 subjects aged 5–70 years (mean age 36.3) including the offspring of the original cohort and the spouses of the offspring were recruited. The offspring cohort has been examined every four years (except the first two examinations with eight years intervening). Within the study, the 330 largest extended families were selected and genotyped with a 10 cm density microsatellite marker and the Affymetrix Human GeneChip 100 K genome-wide scans. The number of subjects with microsatellite genotyped was 1702 and with the 100 K SNPs genotyped was 1343. A recent study reported no evidence of major population substructure in the Framingham Heart Study [8]. LDH was measured during the first (1971–1975) and second (1980–1983) examinations of the offspring. Our analysis was limited to the first examination, since far fewer individuals have serum LDH measured in the second examination.
Total LDH was measured by kinetic method using Dow UV LDH-Reagent Kit. Weight was measured with the subject in light clothing and shoes off. Laboratory measurements were made on 12 hours fasting venous blood samples that were collected in tubes containing 0.1% EDTA. Lipid determinations were performed at the Framingham Heart Study laboratory, which participates in the Standardization Program of the Centers for Disease Control. All subjects provided informed consent prior to each clinic visit, and the examination protocol was approved by the Institutional Review Board at Boston Medical Center (Boston, MA). The clinical and laboratory methods have been detailed elsewhere [5].
2.1. Genotyping Methods
For the microsatellite genotyping, genomic DNA was isolated from nucleated blood cells. DNA samples were sent to the Marshfield Mammalian Genotyping Service (http://research.marshfieldclinic.org/genetics/). At an average of 10 cm density, 399 microsatellite markers (Screening Set 9) [9] covered the genome with an average marker heterozygosity of 0.77. The genotyping data were cleaned in two steps. First, the sib-kin program in Aspex (ftp://lahmed.standord.edu/pub/aspex/index.html) was used to verify family relationships based on all markers available. Second, the GENTEST program, as a precursor of INFER, created by Southwest Foundation for Biomedical Research (http://www.sfbr.org/sfbr/public/software/software.html), was used to identify and eliminate additional genotype inconsistencies. When inconsistencies were found, the genotyping values in all members of the nuclear family were set to missing.
For the Affymetrix 100 K SNPs genotyping, the details are provided in [10]. In summary, SNPs on the Affymetrix Human GeneChip 100 K (n = 112,990 autosomal SNPs) were genotyped in a sample of 1343 individuals from 330 families. SNPs with minor allele frequency <10% or call rate <80% or Hardy-Weinberg equilibrium P-value < .001 were excluded, leaving 70591 SNPs for analysis, criteria set by the Framingham Heart Study for the Affymetrix 100 K SNPs association genome-wide scans [11]. The 100 K SNPs data are publicly available through dbGaP (http://www.ncbi.nlm.nih.gov/gap) under Framingham SHARe project.
2.2. Statistical Methods
2.2.1. Genome-Wide Linkage Analysis
Variation in LDH from known factors was identified and removed by regression modeling incorporated in SOLAR [12–14], to enhance the ability of linkage analysis to detect genetically determined variation using a maximum-likelihood-based variance decomposition method. The covariates selected (P < .05) and incorporated into both the heritability estimation and the linkage analyses were age, sex, height, weight, high density lipoprotein cholesterol (HDL-C), and alkaline phosphatase (ALP).
An estimate of heritability was obtained using the variance-component method. Heritability is the proportion of total phenotypic variation due to additive genetic effects, after removing the variation attributable to covariates. The variance component method analysis was also used for the linkage analysis of LDH, adjusted for known covariates, using the random microsatellite DNA markers covering the entire genome. Marker allele frequencies were estimated from the study participants and then used to estimate the proportion of a single-point alleles shared identical by descent (IBD) among all relative pairs. Multipoint IBDs were estimated based on the singlepoint estimates by a regression approach [13]. A likelihood ratio test was used to evaluate linkage by comparing a purely polygenic model (without consideration of genetic marker information) to a model that incorporates IBD information at the marker. The LOD score was the log (base 10) of the ratio of the likelihoods of two models, purely polygenic versus one that also included IBD information at the marker.
Since the variance-component method is based on the assumption of a multivariate normal distribution, violations of this assumption may result in inaccurate results [14–16]. We found that LDH had high kurtosis and thus used an LOD score adjustment method implemented in SOLAR to ensure more reliable results. A fully informative marker linked to the trait studied was simulated. The IBD information for this marker was calculated, and then linkage analysis to the trait was performed. The LOD score adjustment process [15, 16] regresses the observed LOD scores from simulation (10,000 replicates) on the expected LOD scores for a multivariate normal trait to obtain a correction factor for the LOD scores from the analyses of the observed trait. This method provides robust LOD scores for data with nonnormal distributions.
2.2.2. Genome-Wide Association Study
One individual from each family was randomly selected for the Hardy-Weinberg equilibrium (HWE) test using a chi-squared statistic
with one degree of freedom. For each SNP, we modeled the log-transformed trait value adjusted for the same covariates as used
in the linkage analysis. Trait values within each pedigree were assumed to be correlated, while they were independent between
pedigrees. A special case of linear mixed effects model (LME) in SAS was used to fit the model with the covariates and calculate
the residual for each individual. The following model was used for family i, 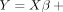 , where Y is a vector of log-transformed LDHs of subjects in the family, and X contains covariates to adjust (age, sex, height, weight, HDL-C, and ALP), which were treated as fixed effects. The covariance
matrix for Y in family i was unstructured. Once the estimate of β was obtained, denoted as
, where Y is a vector of log-transformed LDHs of subjects in the family, and X contains covariates to adjust (age, sex, height, weight, HDL-C, and ALP), which were treated as fixed effects. The covariance
matrix for Y in family i was unstructured. Once the estimate of β was obtained, denoted as 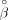 , we calculated the residual for person j in family i with covariates
, we calculated the residual for person j in family i with covariates 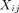 as
as 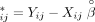 . Then, in the second stage, we modeled the association between the residual and genetic effect by LME given by
. Then, in the second stage, we modeled the association between the residual and genetic effect by LME given by 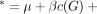 , where the covariance matrix of
, where the covariance matrix of  was unstructured within each family and diagonal between families, and
was unstructured within each family and diagonal between families, and 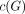 was a coding for the genetic models underlying the disease locus, which were defined as recessive, additive, or dominant.
For each genetic model, a P-value was obtained. The smallest of the three P-values was used to rank all SNPs. In genome-wide association studies, some SNPs associated with common diseases have little
additive effect while there is strong deviation from Hardy-Weinberg equilibrium, which indicates strong recessive or dominant
effects [17, 18]. These SNPs would not be detected using the P-value only based on the additive model. Using the minimum P-value to rank
SNPs will be able to detect SNPs with all three genetic models. Since the minimum P-value is no longer a true p-value, we
used the following threshold values: if the minimum P-value is less than
was a coding for the genetic models underlying the disease locus, which were defined as recessive, additive, or dominant.
For each genetic model, a P-value was obtained. The smallest of the three P-values was used to rank all SNPs. In genome-wide association studies, some SNPs associated with common diseases have little
additive effect while there is strong deviation from Hardy-Weinberg equilibrium, which indicates strong recessive or dominant
effects [17, 18]. These SNPs would not be detected using the P-value only based on the additive model. Using the minimum P-value to rank
SNPs will be able to detect SNPs with all three genetic models. Since the minimum P-value is no longer a true p-value, we
used the following threshold values: if the minimum P-value is less than 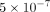 , the association is regarded as strong, while if the minimum P-value is between
, the association is regarded as strong, while if the minimum P-value is between 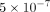 and
and 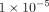 , the association is moderate. No correction for multiple testing was given for the minimum P-value.
, the association is moderate. No correction for multiple testing was given for the minimum P-value.
3. Results
3.1. Genome-Wide Linkage Analysis
The total number of individuals with measured LDH and with all covariates used for the heritability estimates and linkage analysis in offspring was 1603. The basic characteristics of the clinical covariates of these individuals (about 50% male) are displayed in Table 1. Of the 1603 individuals, 1260 had microsatellite marker genotypes.
Characteristics of the 1603 individuals with LDH and covariates used in the linkage analysis.
The skewness and kurtosis of LDH was 1.87 ± 0.04 and 13.92 ± 0.07, respectively. The heritability estimate for LDH, after adjusting for the covariates, was 40.9 ± 6.4%. The proportion of variation due to all covariates included in the model was approximately 5.8%.
From the multipoint linkage analysis of LDH, several maximum LOD scores between 1 and 2.5 were observed on different chromosomes (Table 2). The highest LOD score was 2.41 on chromosome 9.
Chromosomal regions in the genome scan with multipoint LODs ≥ 1.0.
3.2. Genome-Wide Association Study
After log-transformed, only one individual's LDH value, 1.74, was out of the range of 5SD from the mean, 1.77–2.64. Since
the value of 1.74 was so close to the lower bound of 1.77, we did not exclude this individual from the association study and
reanalyzed the data. In the genome-wide association study, no SNP showed strong association, while there were two SNPs, rs9318892
and rs10518949, which showed moderate association. The top ranked SNP on chromosome 13 had a minimum P-value of 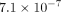 , while the second one on chromosome 15 had a mini p = 1.6 × 10−7. There were 32 SNPs having a minimum P < 10−4, Table 3. All SNPs within 60 kb of LDHA gene (rs4237721, rs2279900, rs8018, rs3781640) and LDHB gene (rs10505873, rs1677106, rs1677104,
rs1030452, rs1012354) have a P-value > .20.
, while the second one on chromosome 15 had a mini p = 1.6 × 10−7. There were 32 SNPs having a minimum P < 10−4, Table 3. All SNPs within 60 kb of LDHA gene (rs4237721, rs2279900, rs8018, rs3781640) and LDHB gene (rs10505873, rs1677106, rs1677104,
rs1030452, rs1012354) have a P-value > .20.
Top 32 SNPs associated with LDH levels with a minimum P-value < 10−4 by LME test.
4. Discussion
In our study, the heritability of LDH was estimated at 41% indicating that a substantial portion of the variation in serum LDH was attributable to additive genetic factors. This is consistent with a previous finding [5]. In the linkage study, we identified several chromosomal regions with LOD scores between 1 and 2.5. None of them are located on the chromosomes where the genes encoding the LDH isoenzymes reside. No obvious candidate genes were found in those chromosomal regions. In the genome-wide association study, no SNP with genome-wide significance was observed. Furthermore, testing three models per SNP in the genome-wise association study adds burden to the multiple test and this is not reflected in the P-values in Table 3. Among the top 32 SNPs with a P-values < 10−4, none of them were near the LDH structure genes, and none were within the regions of linkage peaks. Similarly as in the linkage analysis, no obvious candidate genes were found on those chromosomal regions.
A power study for linkage on the 330 Framingham families with traits measured only in the Offspring Cohort, similar to our
study, was performed using SOLAR. The power was estimated to be 97%, 84%, and 62% to detect a QTL heritability of 30% using
a LOD score cut off of 1, 2, or 3, respectively, as significant (http://www.framinghamheartstudy.org/). The results of the simulation studies imply that this study sample only has sufficient power to detect large QTL effects.
A power study for association on the same study population under additive genetic model demonstrated 97%, 100%, 94%, and 77%
power for SNP minor allele frequencies of 5%, 10%, 20%, and 30%, assuming 10% QTL heritability, the allele frequency for the
QTL to be 0.10, and the QTL and the marker are in linkage disequilibrium (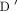 = 1). The power was calculated using PBAT, a software for the family-based association test (the same Framingham website
mentioned above). Usually LME is expected to have higher power than PBAT, since only heterozygous parents are informative
for a family-based study. The results of the simulation studies imply that this study population may have good power for linkage
study. However, since the Affymetrix 100 K GeneChip has a mean marker distance of 26 kb, in most situations, the genomic coverage
may not be high enough to reach the power mentioned above for association study unless there is a major gene with a larger
effect size in linkage disequilibrium with the adjacent SNPs.
= 1). The power was calculated using PBAT, a software for the family-based association test (the same Framingham website
mentioned above). Usually LME is expected to have higher power than PBAT, since only heterozygous parents are informative
for a family-based study. The results of the simulation studies imply that this study population may have good power for linkage
study. However, since the Affymetrix 100 K GeneChip has a mean marker distance of 26 kb, in most situations, the genomic coverage
may not be high enough to reach the power mentioned above for association study unless there is a major gene with a larger
effect size in linkage disequilibrium with the adjacent SNPs.
Although there is a strong genetic effect on the variation of serum LDH levels, the results of both linkage and association genome scans did not show evidence of a major gene effect. Instead, there may be many genes with small effects in controlling the variation of serum LDH levels.
A limitation of this study is that our cohort is mostly composed of Caucasians. Therefore, caution is advised in extrapolating our results to other ethnicities.
Acknowledgments
The authors are very grateful to Nancy Geller for her helpful comments on the data analysis and critical review of the manuscript. This work was supported by the National Heart, Lung, and Blood Institute's Framingham Heart Study (Contract no. N01-HC-25195).
- Received January 29, 2010.
- Revision received May 11, 2010.
- Accepted July 18, 2010.
- © 2010 Jing-Ping Lin et al.















