|
|
Comparaison de schémas thérapeutiques comportant 3 ou 4 antirétroviraux pour le traitement initial de l'infection par le VIH 1Un essai contrôlé randomisé
Roy M. Gulick, MD, MPH;
Heather J. Ribaudo, PhD;
Cecilia M. Shikuma, MD;
Christina Lalama, MS;
Bruce R. Schackman, PhD;
William A. Meyer, III, PhD;
Edward P. Acosta, PharmD;
Jeffrey Schouten, MD, JD;
Kathleen E. Squires, MD;
Christopher D. Pilcher, MD;
Robert L. Murphy, MD;
Susan L. Koletar, MD;
Margrit Carlson, MD;
Richard C. Reichman, MD;
Barbara Bastow, RN, BSN;
Karin L. Klingman, MD;
Daniel R. Kuritzkes, MD; pour le groupe d'étude AIDS Clinical Trials Group (ACTG)
A5095
RÉSUMÉ
| |
Contexte Pour le traitement initial de l'infection par le virus
de l'immunodéficience humaine de type 1 (VIH 1), il est habituel
d'utiliser un schéma thérapeutique comportant 3
antirétroviraux. Un traitement comportant 4 agents pourrait
peut-être cependant améliorer l'activité
antirétrovirale et être plus efficace qu'un traitement
comportant 3 agents.
Objectif Comparer l'efficacité et la
sécurité d'emploi d'un schéma
thérapeutique comportant 3 agents et d'un schéma comportant
4 agents pour le traitement initial de l'infection par le VIH 1.
Plan expérimental L'étude A5095 de l'ACTG
(AIDS Clinical Trials Group) est un essai randomisé, en double
aveugle, contrôlé contre placebo dont le recrutement et le suivi
ont été réalisés entre le 22 mars 2001 et le
1er mars 2005. Les patients, recrutés dans les unités
américaines d'essais cliniques de l'ACTG, devaient être
infectés par le VIH 1, avoir une charge virale d'au moins 400
copies d'ARN-VIH/ml et ne pas avoir déjà été
traités.
Traitements Zidovudine/lamivudine et efavirenz (schéma
à 3 agents) vs zidovudine/0lamivudine/abacavir et efavirenz
(schéma à 4 agents).
Principaux critères de jugement Temps écoulé
jusqu'à l'échec virologique (défini comme le
temps écoulé jusqu'au premier de 2 taux successifs
d'ARN du VIH 1 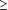 200 copies/ml à la semaine 16 ou
après), modification du nombre de lymphocytes CD4 et
événements indésirables de grade 3 ou 4. Les
données concernant la charge virale ont été
analysées en intention de traiter, quelles que soit les modifications
du traitement. 200 copies/ml à la semaine 16 ou
après), modification du nombre de lymphocytes CD4 et
événements indésirables de grade 3 ou 4. Les
données concernant la charge virale ont été
analysées en intention de traiter, quelles que soit les modifications
du traitement.
Résultats Au total, 765 patients ayant une charge virale
moyenne initiale de 4,86 log (72 444) copies d'ARN-VIH/ml et un nombre
initial moyen de lymphocytes CD4 de 240/mm_ ont été
randomisés. Après un suivi médian de 3 ans, un
échec virologique tel que défini dans le protocole a
été constaté chez 99 patients (26 %) sur 382 dans le
groupe 3 agents et chez 94 patients (25 %) sur 383 dans le groupe 4 agents; le
temps écoulé jusqu'à l'échec virologique
n'était pas significativement différent dans les 2 groupes
(rapport des risques instantanés: 0,95; intervalle de confiance
à 97,5 %: 0,69 — 1,33; p = 0,73). Les analyses planifiées
de sous-groupes ont montré une augmentation du risque
d'échec virologique chez les patients noirs non hispaniques
(rapport des risques instantanés ajusté: 1,66; intervalle de
confiance à 95 %: 1,18 — 2,34; p = 0,003). À 3 ans, dans
le groupe 3 agents vs le groupe 4 agents, la charge virale
était de moins de 200 copies/ml respectivement chez 152 patients (90 %)
sur 169 et chez 143 patients (92 %) sur 156 (p = 0,59) et de moins de 50
copies/ml chez respectivement 144 patients (85 %) sur 169 et chez 137 patients
(88 %) sur 156 (p = 0,39). Aucune différence significative entre
groupes n'a été constatée pour l'augmentation
du nombre de lymphocytes CD4 et pour les événements
indésirables de grade 3 ou 4.
Conclusions Chez des patients n'ayant encore jamais reçu
de traitement, nous n'avons constaté aucune différence
significative entre un schéma thérapeutique
antirétroviral comportant 3 agents et un schéma comportant 4
agents; globalement, la charge virale était inférieure à
50 copies/ml à 3 ans chez au moins 80 % environ des patients. Ces
résultats confirment les recommandations actuelles des guides de bonne
pratique, à savoir d'utiliser 2 nucléosides en association
avec l'efavirenz pour le traitement initial de l'infection à
VIH 1; l'adjonction d'abacavir à ces 3 médicaments
n'a procuré aucun avantage supplémentaire.
Enregistrement des essais cliniques Numéro d'identification
clinicaltrials. gov NCT00013520
JAMA. 2006 ; 296: 769-781.
Le traitement initial habituel de l'infection par le virus de
l'immunodéficience humaine de type 1 (VIH 1) consiste à
utiliser 2 inhibiteurs nucléosidiques de la transcriptase inverse
(INTI) en association avec soit un inhibiteur non nucléosidique de la
transcriptase inverse (INNTI) soit un inhibiteur de protéase
(IP).1,2
Les schémas thérapeutiques antirétroviraux comportant 3
agents permettent d'inhiber la virémie, d'augmenter le nombre
de lymphocytes CD4, de retarder l'évolution clinique et
d'améliorer la
survie.3-6
On a cependant constaté la persistance d'une réplication
virale chez des patients traités dont la virémie était
inhibée.7,8
Certains investigateurs ont suggéré que l'adjonction
d'autres médicaments au schéma thérapeutique
standard pourrait améliorer l'activité
antirétrovirale.9-13
Cependant, ces agents supplémentaires augmentent la complexité,
la toxicité potentielle et les coûts du traitement et, par
ailleurs, les études qui ont comparé des schémas
antirétroviraux à 3 ou 4 agents ont donné des
résultats
discordants.14-16
L'étude A5095 de l'ACTG (AIDS Clinical Trials
Group) avait été conçue au départ pour
comparer, pour le traitement initial de l'infection à VIH 1, 3
schémas thérapeutiques antirétroviraux simples et bien
tolérés: 3 INTI en traitement combiné (schéma
à 3 nucléosides); 2 INTI en traitement combiné et
efavirenz (schéma thérapeutique standard à 3 agents); et
3 INTI en traitement combiné et efavirenz (schéma à 4
agents). Le schéma à 3 nucléosides a été
arrêté rapidement, après 32 semaines de suivi
médian, en raison de son infériorité sur le plan
virologique; les résultats des comparaisons entre les données du
groupe 3 nucléosides et les données combinées des 2
autres groupes ont été analysés et
publiés.17
Il était néanmoins toujours intéressant de savoir si le
schéma comportant 4 agents aurait une activité
antirétrovirale supérieure à celle du schéma
standard comportant 3 agents. Afin d'étudier cette question, nous
avons poursuivi, durant la période médiane de 3 ans
planifiée au départ, une surveillance en aveugle des
participants de l'étude ACTG A5095 qui recevaient un schéma
thérapeutique, à 3 ou à 4 agents, comportant de
l'efavirenz; ce sont ces résultats que nous rapportons ici.
MÉTHODES
Participants
Les participants potentiels à l'étude ont
été recrutés et ont subi une évaluation dans 33
unités de l'ACTG. Les patients ont indiqué eux-mêmes
leur race/ethnie (Amérindiens/Autochtones de l'Alaska;
Asiatiques/Natifs des îles du Pacifique; Noirs, non Hispaniques;
Hispaniques [quelle que soit la race]; Blancs, non Hispaniques; Autres/race
inconnue). Pour participer à l'étude, les patients devaient
être des adultes infectés par le VIH 1, n'ayant pris aucun
traitement antirétroviral au préalable et ayant une charge
virale plasmatique 400 copies d'ARN-VIH/ml (Test Amplicor ou
UltraSensitive HIV-1 Monitor version 1.0; Roche Molecular Systems, Branchburg,
New Jersey). Les patients étaient exclus s'ils avaient reçu
un immunomodulateur, un agent expérimental ou un vaccin dans les 30
jours précédant l'entrée dans l'étude,
s'ils pesaient moins de 40 kg ou s'il s'agissait d'une
femme enceinte ou allaitant.
Plan expérimental
L'étude ACTG A5095 avait été conçue au
départ comme un essai randomisé, en double aveugle,
contrôlé contre placebo, évaluant 3 schémas
antirétroviraux de traitement de l'infection à VIH 1. Elle
devait recruter 1125 patients infectés par le VIH 1, ceux-ci ayant une
probabilité identique de recevoir par randomisation un des 3
schémas thérapeutiques par voie orale suivants: schéma
à 3 nucléosides (zidovudine/lamivudine/abacavir, en traitement
combiné comportant respectivement 300/150/300 mg de chaque agent
[Trizivir, GlaxoSmithKline, Research Triangle Park, Caroline du Nord] pris
oralement 2 fois par jour); schéma thérapeutique standard
à 3 agents (zidovudine/lamivudine, en traitement combiné
constitué de 300/150 mg [Combivir, GlaxoSmithKline] pris oralement 2
fois par jour, et efavirenz, 3 comprimés à 200 mg [Sustiva,
Bristol-Myers Squibb, New York] pris 1 fois par jour); schéma à
4 agents (zidovudine/lamivudine/abacavir [traitement combiné
constitué de 300/150/300 mg, pris 2 fois par jour] et efavirenz [3
comprimés à 200 mg pris 1 fois par jour]). Au départ,
tous les schémas thérapeutiques comportaient 7 comprimés
(y compris les différents placebos) répartis sur 2 prises
quotidiennes, puis, après l'apparition des comprimés
à 600 mg d'efavirenz, ils ne comportèrent plus que 5
comprimés répartis sur 2 prises quotidiennes. La randomisation a
été stratifiée en fonction de la charge virale
constatée lors du dépistage (< 100 000 copies/ml ou
100000 copies/ml).
Des consultations ont été programmées pour
l'étude aux semaines 2, 4, 8, 12, 16, 20 et 24 puis toutes les 8
semaines et ont été maintenues même en cas de modification
ou d'arrêt du traitement. Une évaluation clinique
était effectuée à chaque consultation, ainsi que des
examens biologiques, en particulier l'évaluation de la charge
virale plasmatique (Test HIV-1 Monitor, version 1.0 jusqu'en avril 2003
puis version 1.5) réalisée de manière centralisée
dans le laboratoire de référence du Johns Hopkins Hospital,
Baltimore, Maryland. Une numération des lymphocytes CD4 a
été réalisée lors des consultations des semaines 4
et 8 puis toutes les 8 semaines dans un laboratoire répondant aux
normes CLIA (Clinical Laboratory Improvement Amendments) ou un
équivalent ayant adhéré au programme d'assurance
qualité en immunologie de l'ACTG. Une recherche
d'antigène de surface du virus de l'hépatite B (Ag
HBs) et d'anticorps anti-virus de l'hépatite C (AC anti-VHC)
a également été demandée au départ de
l'étude. L'observance thérapeutique a
été évaluée aux semaines 4, 12 et 24 puis toutes
les 24 semaines au moyen d'un
questionnaire18
rempli par les participants, seuls ou aidés d'un coordinateur de
l'étude.
Si la toxicité d'un des médicaments étudié
représentait un obstacle à la poursuite du traitement,
l'investigateur sur site pouvait substituer la stavudine (Zerit,
Bristol-Myers Squibb) à la zidovudine, la didanosine (Videx EC,
Bristol-Myers Squibb) à l'abacavir et/ou la névirapine
(Viramune; Boehringer-Ingelheim, Ingelheim, Allemagne) à
l'efavirenz; dans ce cas, le patient était tout de même
considéré comme recevant le schéma thérapeutique
initial. En cas d'échec virologique, le patient pouvait recevoir
un autre médicament antirétroviral fourni par
l'équipe de l'étude et choisi en fonction des
résultats d'un test génotypique de résistance
médicamenteuse (HIV-1 TRUGENE; Bayer Healthcare Diagnostics, Berkeley,
Californie). Ce test n'était réalisé que si la
charge virale était supérieure à 500 copies/ml, la
probabilité d'obtenir un résultat étant trop faible
dans le cas contraire. Les événements indésirables
étaient évalués par les équipes sur site, un grade
leur étant attribué en utilisant l'échelle de
toxicité du département SIDA du National Institute of
Allergy and Infectious
Diseases.19
Le protocole d'étude et toutes ses modifications
ultérieures ont été soumis aux comités
d'éthique des différents sites qui les ont examinés
et approuvés. Les équipes sur site ont revu les objectifs de
l'étude, son plan expérimental, ses procédures, ses
risques, ses bénéfices potentiels et les solutions offertes aux
patients ne souhaitant pas participer. Les participants ont signé un
formulaire de consentement éclairé avant toute procédure
liée à l'étude et pour chaque modification du
protocole. Les participants étaient en outre informés que la
confidentialité des renseignements les concernant serait assurée
et qu'ils ne pourraient être identifiés personnellement dans
les publications concernant l'étude.
Historique de l'étude
Le recrutement a eu lieu entre le 22 mars 2001 et le 4 novembre 2002. A la
suite de sa deuxième évaluation annuelle, le 6 février
2003, le comité de surveillance des données du National
Institute of Allergy and Infectious Diseases a conclu que le
schéma comportant 3 nucléosides était inférieur
sur le plan virologique à chacun des 2 schémas contenant de
l'efavirenz et que, par conséquent, les critères
d'arrêt de l'étude spécifiés dans le
protocole étaient remplis. Le comité a recommandé de
révéler le traitement pris par les participants du groupe 3
nucléosides et d'arrêter ce traitement. Il a
également demandé que soient communiqués les
résultats des comparaisons entre les données de ce groupe et les
données combinées des 2 autres groupes; ces résultats,
obtenus après un suivi médian de 32 semaines, ont
été analysés et
publiés.17
Le suivi en aveugle a été poursuivi pour les 2 groupes ayant
un schéma thérapeutique comportant de l'efavirenz. Il
était prévu au départ que le suivi se termine 96 semaines
après la randomisation du dernier patient. Lors de sa troisième
évaluation, le 3 février 2004, le comité a
constaté que les taux d'événements étaient
plus faibles que prévu et il a, par conséquent, autorisé
24 semaines de suivi supplémentaire, la fin de l'étude
étant programmée pour le 1er mars 2005, avec un suivi
médian de 144 semaines, et l'analyse des données
commençant juste après.
Méthodes statistiques
Le principal critère de jugement de la sécurité
d'emploi était le temps écoulé jusqu'à
la survenue du premier événement indésirable de grade 3
ou 4. Le principal critère de jugement de l'efficacité
était le temps écoulé jusqu'à
l'échec virologique, défini comme le temps
écoulé jusqu'au premier de 2 taux successifs d'ARN du
VIH 1 supérieurs ou égaux à 200 copies/ml à la
semaine 16 ou après; les distributions des temps écoulés
jusqu'à l'échec virologique des 2 groupes ont
été comparées. L'étude a été
conçue pour avoir une puissance de 80 % de détecter un rapport
des risques instantanés (RRI) de 0,70 pour l'échec
virologique entre les différents schémas.
Toutes les valeurs de p et tous les intervalles de confiance (IC)
indiqués sont théoriques, non corrigés pour comparaisons
et analyses intérimaires multiples. Dans la mesure où toutes les
comparaisons 2 à 2 entre les 3 groupes d'étude
d'origine avaient été planifiées, les comparaisons
entre les groupes 3 agents et 4 agents et les comparaisons secondaires ont
été évaluées avec un seuil de signification de 2,5
% et des intervalles de confiance à 97,5 % afin de préserver le
risque de première espèce global. Pour les RRI, le
numérateur est le groupe 4 agents. En l'absence d'indication
contraire, les analyses de l'efficacité sont en intention de
traiter sans tenir compte d'un éventuel arrêt du traitement
et les données manquantes sont ignorées; cette hypothèse
a été évaluée en utilisant un modèle de
régression logistique pour données à mesures
répétées.20
Des analyses des patients sous traitement et des analyses de
sensibilité assimilant l'arrêt du traitement, les
données manquantes ou les 2 à des échecs ont
été utilisées pour examiner la solidité des
conclusions de l'étude faites avec ces hypothèses
principales. Les analyses de la sécurité d'emploi ont
été faites sur la population sous traitement
étudié, en incluant uniquement (en conformité avec la
convention de l'ACTG) les patients ayant commencé un des
traitements étudiés et ayant un suivi de 8 semaines après
arrêt du traitement. La charge virale initiale et le nombre initial de
CD4 ont été déterminés comme, respectivement, la
moyenne géométrique et la moyenne arithmétique des
évaluations faites avant l'entrée et à
l'entrée dans l'étude.
Des méthodes prenant en compte le temps écoulé
jusqu'à l'échec ont été utilisées
pour évaluer les objectifs de sécurité d'emploi et
d'efficacité. Les distributions des temps écoulés
jusqu'à l'échec ont été estimées
en utilisant la méthode de Kaplan Meier et comparées par test
des rangs logarithmiques stratifié. Des modèles des risques
proportionnels de Cox, avec stratification sur la charge virale
constatée lors du dépistage, ont été
utilisés pour l'estimation des RRI et des IC. Les interactions
entre les covariables ont été étudiées dans ces
modèles et la validité de l'hypothèse des risques
proportionnels dans ces modèles a été confirmée en
utilisant la méthode de Grambsch et
Therneau.21 Les
proportions, au cours du temps, de patients de chaque groupe ayant une charge
virale inférieure à 200 copies/ml ou inférieure à
50 copies/ml sont indiquées. Des tests de 2 ont
été utilisés pour comparer les groupes à certains
moments spécifiques du suivi. 2 ont
été utilisés pour comparer les groupes à certains
moments spécifiques du suivi.
Pour les nombres de lymphocytes CD4, nous indiquons les changements moyens
estimés par rapport aux valeurs initiales. Des tests t de
Student ont été utilisés pour comparer les groupes
à certains moments spécifiques du suivi. Pour l'apparition
de mutations entraînant une résistance médicamenteuse, des
tests exacts de Fisher ont été utilisés pour les
comparaisons entre groupes. L'observance a été
étudiée comme une covariable dichotomique, en utilisant le
questionnaire habituel de
l'ACTG18 dans
lequel les participants sont considérés avoir une bonne
observance thérapeutique s'ils indiquent n'avoir
oublié aucune prise médicamenteuse durant les 4 jours
précédant l'évaluation.
Toutes les comparaisons entre groupes ont été
évaluées avec le seuil de signification théorique de 2,5
% (p < 0,025) ainsi que nous l'avons indiqué
ci-dessus. Ceci a été fait pour avoir un risque de
première espèce de 5 % sur l'ensemble de
l'étude pour toutes les comparaisons entre groupes. Toutes les
autres valeurs de p (par exemple, celles évaluant les
différences raciales dans les réponses) ont été
évaluées avec un seuil de 0,05. Les logiciels SAS version 8 (SAS
Institute Inc., Cary, Caroline du Nord) et S-plus version 6 (MathSoft Inc.,
Cambridge, Massachusetts) ont été utilisés pour
l'ensemble des analyses.
RÉSULTATS
Caractéristiques initiales
Les caractéristiques initiales des participants sont
résumées dans le
Tableau. Au total, 765
patients se sont vus attribuer par randomisation 1 des 2 schémas
thérapeutiques comportant de l'efavirenz. On comptait parmi les
patients 145 femmes (19 %), 164 sujets hispaniques (21 %), 271 sujets noirs
non hispaniques (35 %) et 314 sujets blancs non hispaniques (41 %). La charge
virale plasmatique moyenne initiale était de 4,86 log (écart
type: 0,73 log) copies d'ARN-VIH/ml (72 444 copies/ml) et le nombre moyen
de lymphocytes CD4 était de 240/mm3 (écart type:
192/mm3). Pour les caractéristiques de départ, les 2
groupes étaient équilibrés.
|
|
|
|
Tableau.. Caractéristiques initiales*.
|
|
|
Répartition des patients
La Figure 1 montre la
répartition des patients dans l'étude. La médiane du
temps de suivi était de 144 semaines (intervalle interquartile: 120-168
semaines). Parmi les 765 patients randomisés, 594 (78 %) ont
achevé l'étude, 157 (20 %) l'ont arrêtée
prématurément et 16 (2 %) sont morts (2 après arrêt
prématuré de l'étude). Les temps
écoulés jusqu'à l'arrêt
prématuré de l'étude n'étaient pas
significativement différents dans les 2 groupes (p = 0,56).
Parmi les 765 patients randomisés, 7 (1 %) n'ont pas
démarré le traitement étudié. A la fin de
l'étude, sur les 758 patients qui avaient démarré le
traitement, 463 (61 %) prenaient toujours le traitement initial, 70 (9 %)
avaient arrêté le traitement initial et prenaient un autre
schéma fourni par l'équipe de l'étude, 61 (8 %)
avaient arrêté le traitement étudié mais
étaient toujours suivis, en prenant ou non un traitement
antirétroviral, et 103 (14 %) avaient arrêté le traitement
étudié (arrêts prématurés de
l'étude, perdus de vue et décès). Globalement, sur
les 295 arrêts de traitement, 79 (27 %) avaient pour motif un
échec virologique et 66 (23 %) un effet indésirable. Les taux
d'arrêt du traitement initial dans les 2 groupes
n'étaient pas significativement différents (p =
0,45).
|
|
|
|
Figure 1.. Répartition des patients dans l'étude A5095 de
l'AIDS Clinical Trials Group. 3TC = lamivudine; ABC = abacavir;
EFV = efavirenz; ZDV = zidovudine. * Détails
rapportés par Gulick et
coll.17
|
|
|
Réponses virologiques et immunologiques
Plus de 90 % des données étaient disponibles
jusqu'à la semaine 192 dans le groupe 3 agents et
jusqu'à la semaine 144 dans le groupe 4 agents. Le groupe 4 agents
étant pris comme numérateur, il n'y avait aucune
différence apparente dans le risque de données manquantes entre
les 2 groupes (odds ratio: 1,05; IC à 97,5 %: 0,73 – 1,53;
p = 0,51 et, en prenant en compte les données manquantes et
les données d'étude arrêtée, odds ratio: 1,14;
IC à 97,5 %: 0,88 – 1,50; p = 0,31).
Globalement, 99 patients (26 %) sur les 382 du groupe 3 agents et 94
patients (25 %) sur les 383 du groupe 4 agents ont eu un échec
virologique tel que défini dans le protocole; les temps
écoulés jusqu'au premier échec virologique
n'étaient pas significativement différents dans les 2
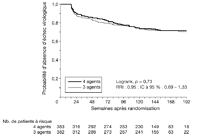
groupes (p = 0,73) (voir Figure
2), avec un RRI estimé de 0,95 (IC à 97,5 %:
0,69 – 1,33). Des résultats similaires ont été
constatés dans des sous-groupes: chez les 437 patients dont la charge
virale était inférieure à 100000 copies/ml au
dépistage, 55 (25 %) sur les 218 participants du groupe 3 agents et 50
(23 %) sur les 219 participants du groupe 4 agents ont eu un échec
virologique; et chez les 328 patients dont la charge virale était
supérieure ou égale à 100000 copies/ml au
dépistage, 44 (27 %) sur les 164 du groupe 3 agents et 44 (27 %) sur
les 164 du groupe 4 agents ont eu un échec virologique. De plus, 14
autres patients (7 dans chaque groupe) ont eu un unique taux, non
confirmé, d'ARN du VIH 1 200 copies/ml à la semaine 16
ou après, avant d'être perdus de vue ou d'abandonner
l'étude. En incluant ces patients dans l'analyse des
échecs virologiques, on obtenait des résultats similaires. Les
analyses de sensibilité prenant d'autres définitions de
l'échec virologique (indiquées ci-dessus) et les analyses
des patients sous traitement initial donnaient également des
résultats similaires.
|
|
|
|
Figure 2.. Temps écoulé jusqu'au premier échec virologique
(analyse en intention de traiter) – Groupe 3 agents vs groupe 4
agents. Le groupe 4 agents a reçu zidovudine/lamivudine/abacavir et
efavirenz; le groupe 3 agents a reçu zidovudine/lamivudine et
efavirenz. Les nombres de patients à risque au cours du temps
représentent les nombres de patients toujours dans l'étude
et n'ayant pas encore eu d'échec virologique (quelle que soit
la situation par rapport au traitement initial); les patients ayant
abandonné l'étude avant un échec virologique sont
censurés au moment de leur dernière évaluation de la
charge virale. Les valeurs de p et les intervalles de confiance
indiqués sont théoriques. IC = intervalle de confiance; Logrank
= test des rangs logarithmiques; RRI = rapport des risques
instantanés.
|
|
|
Les résultats restaient également cohérents
après ajustement sur le sexe, l'âge, la race/ethnie, les
résultats des recherches d'Ag HBs et AC anti-VHC, la charge virale
initiale et le nombre initial de CD4 (RRI ajusté: 0,91; IC à
97,5 %: 0,65 – 1,28). Sur les 193 patients ayant un échec
virologique, 136 (70 %) prenaient le traitement initial, 11 (6 %) avaient
cessé temporairement le traitement initial, 10 (5 %) avaient
arrêté le traitement étudié mais prenaient
d'autres médicaments antirétroviraux (choisis pour chaque
individu par son médecin de premier recours) et 36 (19 %) avaient
arrêté tout traitement. Au moment de la censure des
données, 515 patients (90 %) sur les 572 pour lesquels on n'avait
pas observé d'échec virologique prenaient toujours ou
avaient arrêté temporairement le traitement initial et 57 (10 %)
avaient arrêté totalement le traitement de l'étude
(34 avaient arrêté tout médicament antirétroviral
et 23 prenaient d'autres médicaments antirétroviraux
choisis individuellement par le médecin de premier recours).
Avec le modèle multivariable des risques proportionnels de Cox
ajusté sur le sexe, l'âge, la race/ethnie, les
résultats des recherches d'Ag HBs et AC anti-VHC, la charge virale
initiale et le nombre initial de CD4 et stratifié par groupe de
traitement, on pouvait constater une association entre le risque
d'échec virologique et la race/ethnie (p = 0,03). Il
existait plus exactement une augmentation du risque d'échec
virologique chez les patients noirs non hispaniques par comparaison avec les
patients blancs non hispaniques (RRI ajusté: 1,66; IC à 95 %:
1,18 – 2,34; p = 0,003). Concernant le temps
écoulé jusqu'à l'échec virologique, les
comparaisons entre patients hispaniques ou d'autre race/ethnie et les
patients blancs non hispaniques ne montraient pas de différences
significatives (p > 0,20 pour toutes les comparaisons). Il y avait
également une très petite augmentation du risque
d'échec virologique chez les patients ayant une recherche
d'AC anti-VHC positive (RRI estimé: 1,57; IC à 95 %: 1,02
– 2,41; p = 0,04). Il n'y avait aucune autre association
significative avec le temps écoulé jusqu'à
l'échec virologique pour d'autres covariables initiales
(p > 0,20 dans tous les cas).
La médiane du temps écoulé jusqu'à la
réponse virologique (taux confirmé d'ARN du VHS 1 < 200
copies/ml), quelles que soient les modifications du traitement, était
de 8 semaines dans les 2 groupes (p = 0,64). Les proportions de
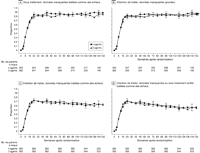
patients ayant moins de 200 copies d'ARN-VIH/ml atteignaient un plateau
à la semaine 16, restant alors constantes autour de 85 % à 90 %
(voir Figure 3A). Les
proportions de patients ayant moins de 50 copies d'ARN-VIH/ml
atteignaient un plateau à la semaine 24, restant alors constantes
autour de 80 % à 85 % (voir
Figure 3B). Les
comparaisons entre groupes réalisées à différents
moments spécifiés au départ ne montraient aucune
différence significative (p < 0,05). Des analyses
complémentaires utilisant différentes approches (intention de
traiter avec données manquantes traitées comme des
échecs, intention de traiter avec données manquantes ou
arrêts de traitement traités comme des échecs, patients
sous traitement initial) ont donné des résultats comparables
(voir Figure 4).
|
|
|
|
Figure 3.. Proportions de patients ayant moins de 200 copies et moins de 50 copies
d'ARN du VIH 1 (analyse en intention de traiter) – Groupe 3 agents
vs groupe 4 agents. Le groupe 4 agents a reçu
zidovudine/lamivudine/abacavir et efavirenz; le groupe 3 agents a reçu
zidovudine/lamivudine et efavirenz. Les nombres de patients indiqués
pour chacune des semaines d'étude (les dénominateurs pour
les proportions) représentent les nombres de patients ayant eu une
évaluation de la charge virale cette semaine là; les
numérateurs des proportions sont les nombres de patients ayant moins de
200 ou moins de 50 copies d'ARN du VIH 1/ml (quelle que soit leur
situation par rapport au traitement). Les données manquantes
(évaluations omises, sujets déjà perdus de vue, censures)
ont été ignorées. Les proportions ont été
comparées aux semaines 24, 48, 72, 96 et 144 en utilisant des tests de
2. Les barres d'erreur indiquent les intervalles de
confiance à 97,5 %. Les valeurs de p et les intervalles de confiance
indiqués sont théoriques, non ajustés pour comparaisons
multiples. Les comparaisons effectuées aux différents moments
d'évaluation spécifiés au départ n'ont
pas montré de différences significatives (p > 0,05
pour toutes les comparaisons).
|
|
|
|
|
|
|
Figure 4.. Proportions de patients ayant moins de 50 copies d'ARN du VIH 1 (4
analyses différentes) – Groupe 3 agents vs groupe 4
agents. Le groupe 4 agents a reçu zidovudine/lamivudine/abacavir et
efavirenz; le groupe 3 agents a reçu zidovudine/lamivudine et
efavirenz. Pour chaque type d'analyse, les nombres de patients
indiqués pour chacune des semaines d'étude (les
dénominateurs pour les proportions) représentent les nombres de
patients: A, prenant le traitement initial et ayant eu une évaluation
de la charge virale la semaine considérée; B, ayant eu une
évaluation de la charge virale quelle que soit sa situation concernant
le traitement; et C et D, pour lesquels une évaluation de la charge
virale était attendue en se basant sur la date de randomisation. Les
numérateurs des proportions sont les nombres de patients ayant: A,
moins de 50 copies d'ARN-VIH/ml (données manquantes
traitées comme des échecs); B, moins de 50 copies
d'ARN-VIH/ml (quelle que soit la situation par rapport au traitement,
données manquantes ignorées); C, moins de 50 copies
d'ARN-VIH/ml (quelle que soit la situation par rapport au traitement,
données manquantes traitées comme des échecs); et D,
moins de 50 copies d'ARN-VIH/ml (données manquantes
traitées comme des échecs, arrêts du traitement initial
traités comme des échecs). Les barres d'erreur indiquent
les intervalles de confiance à 97,5 %. Les valeurs de p et les
intervalles de confiance indiqués sont théoriques, non
ajustés pour comparaisons multiples. Les comparaisons effectuées
aux différents moments d'évaluation spécifiés
au départ n'ont pas montré de différences
significatives (p > 0,05 pour toutes les comparaisons).
|
|
|
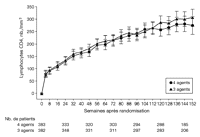
Dans les 2 groupes, les nombres de lymphocytes CD4 ont augmenté
à partir de leurs valeurs initiales (augmentation moyenne globale
à 144 semaines: 291 lymphocytes/mm2; IC à 95 %: 272
– 310) sans différence significative entre groupes (p
> 0,05 quel que soit le moment d'évaluation) (voir
Figure 5).
|
|
|
|
Figure 5.. Nombre de lymphocytes CD4 (modifications à partir des valeurs
initiales) (analyse en intention de traiter) – Groupe 3 agents
vs groupe 4 agents. Le groupe 4 agents a reçu
zidovudine/lamivudine/abacavir et efavirenz; le groupe 3 agents a reçu
zidovudine/lamivudine et efavirenz. Les nombres de patients indiqués
pour chacune des semaines d'étude représentent les nombres
de patients ayant eu une évaluation du nombre de lymphocytes CD4 la
semaine concernée (quelle que soit la situation concernant le
traitement). Les données manquantes (évaluations omises, sujets
déjà perdus de vue, censures) ont été
ignorées. Les changements sont comparés en utilisant des tests t
de Student bilatéraux. Les barres d'erreur indiquent les
intervalles de confiance à 97,5 %. Les valeurs de p et les
intervalles de confiance indiqués sont théoriques, non
ajustés pour comparaisons multiples. Les comparaisons effectuées
aux différents moments d'évaluation spécifiés
au départ n'ont pas montré de différences
significatives (p > 0,05 pour toutes les comparaisons).
|
|
|
Tests génotypiques de résistance
Chez les 193 patients pour lesquels un échec virologique tel que
défini par le protocole d'étude avait été
constaté, des tests génotypiques ont été
effectués rétrospectivement sur les échantillons initiaux
et en temps réel sur les échantillons correspondants à
l'échec virologique. Parmi ces patients, 175 (91 %) avaient au
départ un virus de type sauvage et 18 (9 %) avaient une mutation
associée à une résistance médicamenteuse. Sur ces
18 mutations initiales, il s'agissait dans 7 cas de substitutions
associées à une résistance à 1 seule classe
médicamenteuse (1 à la lamivudine, 1 à un autre INTI, 5
à des INNTI), dans 9 cas de substitutions associées à une
résistance à 2 classes médicamenteuses (4 à des
INTI et des INNTI, 1 à des INTI et des IP et 4 à des INNTI et
des IP) et dans 2 cas de substitutions associées à une
résistance aux 3 classes médicamenteuses. Sur les 193 patients
ayant eu un échec virologique, 15 (8 %) avaient un virus
résistant aux INNTI au départ.
Au moment de l'échec virologique, les résultats du
génotype ont pu être obtenus chez 143 (82 %) des 175 patients
n'ayant pas de résistance initiale, le test génotypique
n'a pas été effectué chez 27 patients (15 %) ayant
une charge virale inférieure à 500 copies/ml et 5 patients (3 %)
n'ont pu avoir de séquençage, le test ne donnant aucun
résultat malgré au moins 2 tentatives. Sur les 143 patients
ayant des résultats de génotype disponibles au moment de
l'échec virologique, 72 (50 %) (37 ayant 3 médicaments et
35 ayant 4 médicaments) avaient un virus de type sauvage; chez les 71
autres patients (50 %) (34 ayant 3 médicaments et 37 ayant 4
médicaments) une mutation de résistance était apparue:
résistance aux INNTI seule (15 patients ayant 3 médicaments et
22 ayant 4 médicaments); M184I/V seule (4 ayant 3 médicaments et
2 ayant 4 médicaments); résistance aux INNTI et M184I/V (13
ayant 3 médicaments et 7 ayant 4 médicaments); M184I/V, autres
mutations associées aux INTI et résistance aux INNTI (1 ayant 3
médicaments et 3 ayant 4 médicaments); M184I/V et autres
mutations associées aux INTI (1 ayant 3 médicaments); et
M184I/V, autres mutations associées aux INTI ou résistance aux
INNTI avec conjointement mutations associées aux IP (3 ayant 4
médicaments).
Parmi les mutations apparues, les mutations significatives associées
à la
zidovudine22 ont
été rares (< 2 %); les substitutions K65R et L74V
conférant une résistance à l'abacavir sont survenues
respectivement 1 fois et pas du tout. Si des résistances à la
lamivudine, aux INNTI ou les 2 ont donc été couramment
sélectionnés au moment de l'échec virologique,
à la fois avec le schéma à 3 médicaments et le
schéma à 4 médicaments, les résistances à
la zidovudine (ou à la stavudine) et à l'abacavir (ou
à la didanosine) ont été rares.
Évaluation de l'observance thérapeutique
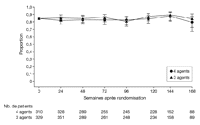
Nous avons obtenu plus de 88 % des évaluations de l'observance
attendues. En ignorant les évaluations manquantes, 82 % à 85 %
des patients indiquaient n'avoir omis aucune prise médicamenteuse
durant les 4 jours précédant l'évaluation, sans
différence significative entre groupes pour chacun des moments
d'évaluation spécifiés au départ (p
> 0,20 dans tous les cas) (voir
Figure 6). Les
résultats étaient similaires quand les données manquantes
étaient considérées comme des prises oubliées: 76
% à 80 % des patients indiquaient n'avoir oublié aucune
prise au cours des 4 jours précédant l'évaluation,
sans différence significative entre groupes pour chaque moment
d'évaluation spécifié au départ (p
> 0,10 dans tous les cas).
|
|
|
|
Figure 6.. Proportions, sur l'ensemble de la période d'étude,
de patients ayant indiqué n'avoir oublié aucune prise du
traitement étudié au cours des 4 jours précédents
— Groupe 3 agents vs groupe 4 agents. Le groupe 4 agents a
reçu zidovudine/lamivudine/abacavir et efavirenz; le groupe 3 agents a
reçu zidovudine/lamivudine et efavirenz. Les nombres de patients
indiqués pour chacune des semaines d'étude (les
dénominateurs pour les proportions) représentent les nombres de
patients prenant le traitement initial et ayant eu une évaluation de
l'observance thérapeutique la semaine concernée. Les
numérateurs des proportions sont les nombres de patients ayant
indiqué un respect total de la prescription médicamenteuse
durant les 4 jours précédant l'évaluation. Les
données manquantes (évaluations omises, sujets
déjà perdus de vue, censures) ont été
ignorées. Les barres d'erreur indiquent les intervalles de
confiance à 97,5 %. Les valeurs de p et les intervalles de
confiance indiqués sont théoriques, non ajustés pour
comparaisons multiples. Les comparaisons effectuées aux
différents moments d'évaluation spécifiés au
départ n'ont pas montré de différences
significatives (p > 0,05 pour toutes les comparaisons).
|
|
|
Evénements indésirables
Au total, 166 patients (22 %) sur 758 ont indiqué l'apparition
d'un événement de grade 4; sur l'ensemble des autres
patients, 257 (34 %) sur 758 ont indiqué l'apparition d'un
événement de grade 3. Concernant le temps écoulé
jusqu'au premier événement indésirable de grade 3 ou
4, aucune différence significative n'a été
constatée entre les 2 groupes (p = 0,39). La survenue
d'un effet indésirable a entraîné une modification du
traitement attribué chez un certain nombre de patients: la zidovudine a
été remplacée par la stavudine chez 69 patients (9 %) sur
758, l'abacavir a été remplacé par la didanosine
chez 25 patients (7 %) sur 382 et l'efavirenz a été
remplacé par la névirapine chez 75 patients (10 %) sur 758.
Des réactions d'hypersensibilité présumées
d'origine médicamenteuse ont été décrites
chez 28 patients (7 %) sur les 376 du groupe 3 agents (prenant un placebo
à la place de l'abacavir) et chez 37 patients (10 %) sur les 382
du groupe 4 agents. La plupart (58/65 [89 %]) des réactions
d'hypersensibilité présumées ont été
attribuées par les investigateurs sur site à l'abacavir. Un
cas de syndrome de Stevens-Johnson a été signalé dans le
groupe 4 agents.
Parmi les 765 patients randomisés, 29 (4 %) ont eu au moins un
événement de la catégorie C définissant le SIDA
des Centers for Disease Control and Prevention et 100 (13 %) (53 [14
%] sur 382 dans le groupe 3 agents et 47 [12 %] sur 383 dans le groupe 4
agents) ont indiqué au moins un événement
indésirable associé au VIH 1 (le plus souvent, un zona
localisé [28], une candidose oropharyngée [25], un herpès
cutanéo-muqueux [24] ou une pneumonie bactérienne à
épisode unique [15]). Il y a eu 2 décès parmi les
patients recevant un traitement dans le groupe 3 agents (accident par
véhicule à moteur [1] et carcinome laryngé [1]) et 3
décès dans le groupe 4 agents (encéphalopathie du VIH [1]
et suicide [2]).
Différences raciales
Nous avons entrepris une série d'analyses
complémentaires pour étudier l'échec virologique en
fonction des différentes races/ethnies. Les taux d'inhibition
virale étaient élevés dans tous les groupes raciaux; par
exemple, les proportions de patients ayant moins de 50 copies/ml
d'ARN-VIH à 144 semaines étaient de 91 % (60 sur 66) chez
les sujets hispaniques, de 80 % (85 sur 106) chez les sujets noirs non
hispaniques et de 89 % (130 sur 146) chez les sujets blancs non hispaniques.
Chez les patients noirs non hispaniques comparés aux patients blancs
non hispaniques, le nombre de CD4 était significativement plus bas au
départ (184 vs 246/mm2; p = 0,007), ainsi
que le nombre absolu de polynucléaires neutrophiles (1890 vs
2312/mm?; p < 0,001). Parmi les patients ayant eu un échec
virologique au cours de l'étude, les sujets noirs non hispaniques
avaient significativement moins de mutations de résistance aux INTI au
départ (0/83 [0 %] vs 5/67 [7 %]) et moins de mutations de
résistance aux INNTI au départ (5/83 [6 %] vs 9/67 [13
%]) que les sujets blancs non hispaniques.
Les temps écoulés jusqu'à l'arrêt du
schéma thérapeutique initial comportant de l'efavirenz
étaient significativement plus courts chez les sujets noirs non
hispaniques que chez les sujets blancs non hispaniques (RRI: 1,37; IC à
95 %: 1,03 – 1,84; p = 0,03). Le motif le plus fréquent
d'arrêt du traitement (chez 43 [36 %] sur 118 sujets noirs non
hispaniques et chez 24 [23 %] sur 105 sujets blancs non hispaniques)
était l'échec virologique. Il n'y avait pas de
différence significative dans les temps écoulés
jusqu'à l'arrêt de l'étude (p =
0,22). Chez les sujets noirs non hispaniques, le temps écoulé
jusqu'à l'apparition d'un effet indésirable de
grade 3 ou 4 – une neutropénie principalement –
était plus court que chez les sujets blancs non hispaniques (RRI: 1,38;
IC à 95 %: 1,09 – 1,73; p = 0,007). Il n'y avait
pas de différence significative entre sujets hispaniques et blancs non
hispaniques concernant les temps écoulés jusqu'à
l'arrêt du schéma thérapeutique (p = 0,55),
l'arrêt de l'étude (p = 0,61) ou
l'apparition d'un effet indésirable de grade 3 ou 4
(p = 0,92).
Les patients noirs non hispaniques ont indiqué significativement
plus souvent que les patients blancs non hispaniques avoir omis au moins 1
prise médicamenteuse au cours des 4 jours précédents,
lors des évaluations de la semaine 4 (46 [20 %] sur 231 vs 28
[11 %] sur 255; p = 0,01) et de la semaine 12 (50 [22 %] sur 226
vs 32 [12 %] sur 267; p = 0,01). Les évaluations des
semaines 24 et 48 ne faisaient cependant apparaître aucune
différence d'observance indiquée par les patients
eux-mêmes entre les différents groupes raciaux (p >
0,20 pour toutes les comparaisons). Par ailleurs, en utilisant un
modèle des risques proportionnels de Cox, nous avons constaté
une interaction entre la race et l'observance à la semaine 12
(p = 0,05): le temps écoulé jusqu'à
l'échec virologique était significativement plus court chez
les sujets noirs non hispaniques qui avaient indiqué avoir
oublié au moins 1 dose du schéma thérapeutique comportant
de l'efavirenz dans les 4 jours précédents que chez les
sujets de race blanche non hispanique ayant eu une observance
thérapeutique similaire (p < 0,001 au test des rangs
logarithmiques); cette interaction n'était pas constatée
à la semaine 4 (p = 0,34). Il n'y avait aucune
différence dans les taux d'échec virologique selon la
race/ethnie dans le groupe plus important de patients indiquant n'avoir
oublié aucune prise lors de l'évaluation de la semaine 12
(p = 0,18) (voir Figure
7). La répétition de cette analyse en utilisant
les données des 3 groupes initiaux de traitement, incluant le groupe
à 3
nucléosides17
mais restreint aux sujets noirs non hispaniques et blancs non hispaniques,
suggérait une interaction à 3 facteurs entre le traitement,
l'observance thérapeutique et la race/ethnie (p = 0,04);
bien qu'il y ait eu des arguments en faveur d'une interaction entre
la race et l'observance à la semaine 12 dans les groupes recevant
un traitement contenant de l'efavirenz (p = 0,06), aucune
interaction significative n'était observée avec le
traitement comportant 3 nucléosides (p = 0,41).
|
|
|
|
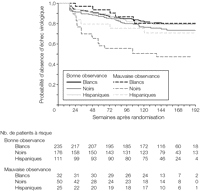
Figure 7.. Temps écoulé jusqu'au premier échec virologique
par race/ethnie et observance indiquée par les participants la semaine
12. Les nombres de patients à risque au cours du temps
représentent les nombres de patients ayant eu une évaluation de
l'observance thérapeutique à 12 semaines et présents
dans l'étude sans avoir eu d'échec virologique (quelle
que soit la situation par rapport au traitement initial); les patients sortis
précédemment de l'étude avant un échec
virologique ont été censurés au moment de leur
dernière évaluation de la charge virale. Les patients
n'ayant pas eu d'évaluation de l'observance la semaine
12 ont été exclus de l'analyse. La mention "mauvaise
observance" signifie que les patients ont indiqué avoir
oublié au moins 1 dose du traitement étudié au cours des
4 jours précédant la consultation. La mention "bonne
observance" signifie que les patients ont indiqué n'avoir
oublié aucune dose durant les 4 jours précédant la
consultation.
|
|
|
COMMENTAIRES
Nous n'avons constaté, sur une période
d'étude de 3 ans, aucune différence significative entre un
schéma thérapeutique standard à 3 agents comportant
zidovudine/lamivudine et efavirenz et un schéma thérapeutique
à 4 agents comportant zidovudine/lamivudine/abacavir et efavirenz en
traitement initial de l'infection à VIH 1, que ce soit pour la
réponse virologique initiale, le temps écoulé
jusqu'à l'échec virologique, le nombre de lymphocytes
CD4, les événements indésirables, l'observance
thérapeutique, les mutations de résistance médicamenteuse
constatées au moment de l'échec virologique,
l'arrêt du traitement ou l'arrêt de l'étude.
Nous avons indiqué dans une publication antérieure que les
résultats obtenus avec un schéma thérapeutique comportant
3 nucléosides étaient inférieurs sur le plan virologique
aux résultats combinés de ces 2 schémas
thérapeutiques comportant de l'efavirenz après une
période médiane de suivi de 32
semaines.17
Globalement, les résultats obtenus chez les patients prenant un
schéma thérapeutique contenant de l'efavirenz sont bons,
avec au moins 80 % environ des patients ayant une diminution de la charge
virale à moins de 50 copies d'ARN-VIH/ml après 3 ans (144
semaines) de suivi. Au cours des 3 ans d'étude, 155 patients (20
%) ont été perdus de vue (dont 104 patients [14 %] perdus de vue
avant constatation du principal critère de jugement de
l'étude); ce taux de suivi est similaire à ceux
constatés dans d'autres essais cliniques comparables
effectués chez des patients infectés par le VIH 1 et non
traités
auparavant.14,23,24
Bien que l'on ait suggéré que l'adjonction
d'un quatrième médicament au schéma
thérapeutique habituel à 3 agents prescrit en traitement initial
de l'infection à VIH 1 puisse améliorer le devenir du
patient,
9-13
les essais effectués jusqu'à présent ont
donné des résultats inégaux, les mauvais résultats
provenant généralement de l'utilisation de schémas
thérapeutiques peu pratiques et nocifs comportant INNTI et IP
associés.14-16
Dans l'étude ACTG 388, par exemple, dans laquelle des patients
avaient reçu par randomisation soit un schéma à 3 agents
(zidovudine/lamivudine et indinavir) soit l'un de 2 schémas
thérapeutiques à 4 agents (zidovudine/lamivudine plus indinavir
et efavirenz; zidovudine/lamivudine plus indinavir et nelfinavir), la
réponse virologique a été meilleure avec le schéma
à 4 agents contenant de l'efavirenz qu'avec le schéma
à 3 agents mais elle a été moins bonne avec le
schéma à 4 agents comportant du nelfinavir qui présentait
par ailleurs une toxicité plus
élevée.14
Dans l'étude ACTG 384 dans laquelle étaient comparés
des schémas thérapeutiques séquentiels à 3 agents
et des schémas à 4 agents, aucune différence
significative de réponse virologique ou de toxicité n'a
été constatée entre les groupes zidovudine/lamivudine
plus soit nelfinavir soit efavirenz et le groupe zidovudine/lamivudine plus
efavirenz et
nelfinavir.15 Dans
l'étude INITIO,
16 les patients ont
reçu par randomisation soit l'un des 2 schémas à 3
agents (stavudine et didanosine plus soit efavirenz soit nelfinavir) soit un
schéma à 4 agents (stavudine et didanosine plus nelfinavir et
efavirenz); la réponse virologique a été meilleure avec
le schéma à 3 agents comportant de l'efavirenz sans
différence de toxicité. Notre étude, qui a comparé
des schémas thérapeutiques pratiques et bien
tolérés, n'a montré aucun avantage
supplémentaire du traitement comportant 4 agents sur le traitement
standard à 3 agents.
Nous avons également constaté dans notre étude que,
bien que les taux de réponse virologique aient été
élevés dans tous les groupes ethniques/raciaux, le temps
écoulé jusqu'à l'échec virologique
était significativement plus court chez les sujets noirs non
hispaniques (mais pas chez les sujets hispaniques) que chez les sujets blancs
non hispaniques. Aucune différence de réponse virologique selon
le groupe racial n'avait été initialement décrite
dans les essais cliniques de phase III portant sur des schémas
thérapeutiques comportant de l'efavirenz; il faut toutefois noter
que peu de sujets noirs et hispaniques étaient
représentés dans ces
essais.23,25
Une analyse complémentaire d'1 de ces essais, effectuée
chez 411 patients (22 % de sujets noirs et 17 % d'Hispaniques) prenant un
traitement de zidovudine/lamivudine plus efavirenz, avait montré que le
temps écoulé jusqu'à l'échec virologique
était plus court chez les Hispaniques que chez les sujets
blancs.26 Dans
l'étude ACTG 384, les sujets noirs et hispaniques étaient
mieux représentés mais aucune différence raciale dans la
réponse au schéma comportant de l'efavirenz n'avait
été
constatée.15,27
Il y a eu cependant au moins 2 publications à avoir noté une
différence raciale de réponse virologique. Dans une étude
rétrospective portant sur 450 patients (dont la majorité
était de race noire), on a constaté que le temps
écoulé jusqu'à l'échec virologique
était significativement plus court chez les sujets noirs prenant un
traitement contenant de l'efavirenz que chez les sujets blancs; il
n'y avait par contre aucune différence avec les schémas
thérapeutiques à base d'indinavir ou de
nelfinavir.28 En
outre, des investigateurs du Johns Hopkins Hospital ont constaté, chez
283 patients du centre hospitalier recevant un traitement comportant de
l'efavirenz (77 % d'Afro-américains et 23 % de sujets blancs
non hispaniques), que la probabilité d'arrêt du traitement
avant 1 an était significativement plus élevée chez les
Afro-américains (32 %) que chez les sujets blancs (16 %) (p =
0,002) et que les taux d'abaissement de la charge virale à moins
de 400 copies d'ARN-VIH/ml étaient plus faibles chez les
Afro-américains (66 % vs 82 %; p =
0,01).29 Par
contre, aucune différence significative n'était
constatée pour les taux d'arrêt du traitement et
d'inhibition virologique chez les patients de cette étude qui
avaient reçu un traitement à base d'IP, mais il n'y
avait que 85 patients concernés (81 % d'Afro-américains et
19 % de sujets blancs non hispaniques).
Nous avons recruté dans notre importante étude 35 % de sujets
noirs non hispaniques et 21 % d'Hispaniques. Le nombre absolu de
polynucléaires neutrophiles était significativement plus bas au
départ chez les sujets noirs non hispaniques que chez les sujets blancs
non hispaniques, le temps écoulé avant la survenue d'un
premier événement indésirable de grade 3 ou 4 – une
neutropénie principalement – étant par conséquent
plus court. On a décrit l'existence de neutropénies
bénignes chez les individus de race
noire.30 Il est
important de noter que les taux d'arrêt de l'étude
n'étaient pas significativement différents dans les
différents groupes ethniques/raciaux et qu'il n'y avait pas
de différence dans l'observance thérapeutique
indiquée par les participants aux semaines 24 et 48 heures, le taux
d'observance indiquée aux semaines 4 et 12 étant cependant
plus faible d'environ 10 % chez les sujets noirs non hispaniques que chez
les sujets blancs non hispaniques. Nous avons aussi constaté une
interaction significative entre la race et l'observance
thérapeutique: le temps écoulé jusqu'à
l'échec virologique était plus court chez les sujets noirs
non hispaniques ayant indiqué avoir oublié au moins 1 dose de
leur traitement contenant de l'efavirenz au cours des 4 jours
précédant l'évaluation de la semaine 12 que chez les
sujets blancs non hispaniques ayant également eu une mauvaise
observance thérapeutique. Aucune différence ethnique/raciale
n'a été observée pour les patients ayant
indiqué une bonne observance thérapeutique lors de
l'évaluation de la semaine 12. Il faut cependant noter que le
nombre de sujets dans certains groupes était faible, ce qui limitait
nos possibilités de déductions. Nous n'avons
constaté aucune interaction entre la mauvaise observance
thérapeutique et la race chez les patients prenant le schéma
à 3 nucléosides mais le temps de suivi était court. Il
faut noter que les patients porteurs d'AC anti-VHC avaient
également des taux d'échec virologique
légèrement mais significativement plus élevés que
les patients n'ayant pas d'AC anti-VHC mais cette constatation
n'expliquait pas les différences ethniques/raciales.
Nous avons rapporté précédemment que la
présence d'un polymorphisme génétique T/T en
position 516 – qui survient plus fréquemment chez les Noirs et
qui code pour CYP2B6, l'enzyme hépatique responsable du
métabolisme de l'efavirenz – est associée à
des taux plasmatiques d'efavirenz significativement plus
élevés et à des symptômes neurologiques centraux
plus précoces, mais nous n'avons pas trouvé de
différences de taux d'arrêt du traitement, de
tolérance ou de réponse virale dans cette analyse
secondaire.31 On
peut raisonnablement envisager la présence de ce polymorphisme
génétique comme une explication possible à nos
constatations actuelles, celui-ci ayant pour conséquence des niveaux
d'efavirenz plus élevés et des effets indésirables
plus marqués conduisant à une moins bonne observance, au
développement d'une résistance au médicament et
finalement à un échec virologique. Il y a d'autres
explications possibles, par exemple des différences ethniques/raciales
qui proviendraient de différences dans des facteurs influençant
l'observance (prise de drogue, effets secondaires légers ou
modérés, soutien social). Ces données sont
limitées par le fait que l'observance est indiquée par les
patients eux-mêmes et que les interactions race/ethnie, observance et
traitement ont été observées lors de
l'évaluation de la semaine 12 mais pas lors de
l'évaluation de la semaine 4. La connaissance des
différences génétiques et ethniques/raciales de
réponse aux médicaments antirétroviraux et de
toxicité de ces médicaments est essentielle dans la mesure
où ces traitements sont recommandés et utilisés dans le
monde entier, dans des populations très
diverses.32
Les traitements à base d'efavirenz de cette étude
étaient associés à une augmentation de presque 300
lymphocytes CD4 par mm2 sur les 3 ans d'études. Bien
que certains aient suggéré que l'augmentation du nombre de
CD4 soit plus importante avec les traitements à base d'INNTI
qu'avec ceux à base d'IP,
33-35
nos résultats semblent comparables à ceux des études
évaluant les IP à long
terme.36
Une résistance aux INNTI a été détectée
rétrospectivement par un test génotypique réalisé
sur un échantillon de départ conservé chez 8 % des
patients qui après avoir débuté un traitement comportant
de l'efavirenz ont eu un échec virologique. Par contre, pour les
81 participants ayant eu un échec virologique alors qu'ils
prenaient un traitement à 3 nucléosides, une résistance
aux INNTI n'a été détectée que dans un seul
échantillon
initial.17 Bien que
ces données suggèrent qu'une résistance
médicamenteuse primaire puisse avoir joué un rôle dans une
petite proportion de cas d'échec thérapeutique, une analyse
des échantillons de départ des participants n'ayant pas eu
d'échec thérapeutique est nécessaire avant de
pouvoir tirer des conclusions. Dans la mesure où les tests de
résistance médicamenteuse n'ont pas été
effectués systématiquement au départ et où, par
conséquent, nous ne disposons pas actuellement de résultats pour
les participants n'ayant pas eu d'échec thérapeutique,
nous ne pouvons tirer de conclusions définitives de ces données;
néanmoins elles suggèrent l'existence d'un risque
d'échec chez les patients ayant une résistance
génotypique initiale et recevant un traitement comportant de
l'efavirenz, ce qui pourrait être un argument en faveur de
l'adoption de nouvelles recommandations indiquant d'évaluer
la résistance médicamenteuse au
départ.1,37
Les effets indésirables constatés dans cette étude ont
été identiques à ceux déjà associés
aux différents médicaments; l'adjonction d'abacavir au
schéma thérapeutique à 3 agents n'a pas
entraîné d'augmentation du nombre
d'événements indésirables. On a montré des
réactions d'hypersensibilité avec
l'efavirenz38
comme avec
l'abacavir.39
La détermination du médicament en cause quand les 2 agents sont
débutés en même temps est un problème difficile
pour le clinicien. Dans cette étude contrôlée contre
placebo, 24 cas de réactions d'hypersensibilité
habituellement associées à l'abacavir ont été
constatés chez des patients prenant le schéma à 3 agents
comportant un placebo d'abacavir. La survenue d'une réaction
hypersensibilité à l'abacavir chez les sujets de race
blanche est associée à la présence de
l'allèle
HLA-B*5701.40
Un dépistage prospectif permettant d'identifier les patients
porteurs de cet allèle a été
proposé.41
CONCLUSIONS
Les résultats de l'étude ACTG A5095 n'ont
montré aucune différence significative, ni pour
l'efficacité ni pour la tolérance, entre un traitement par
zidovudine/lamivudine et efavirenz et un traitement par
zidovudine/lamivudine/abacavir et efavirenz durant une période de suivi
de 3 ans. Les taux élevés d'inhibition virologique
constatés dans cette étude confirment les recommandations
actuelles pour le traitement initial de l'affection à VIH 1,
à savoir utiliser comme schéma thérapeutique
préférentiel l'association de 2 nucléosides et de
l'efavirenz.1,2
L'adjonction d'un quatrième agent, l'abacavir, au
schéma thérapeutique standard à 3 agents n'a
modifié ni la toxicité du traitement ni l'observance
thérapeutique mais n'a apporté aucun avantage
supplémentaire.
Informations sur les auteurs
Correspondance: Roy M. Gulick, MD, MPH, Cornell HIV Clinical Trials
Unit, Division of International Medicine and Infectious Diseases, Weill
Medical College of Cornell University, Box 566, 525 E 68th St, New York, NY
10021
(rgulick{at}med.cornell.edu).
Les affiliations des auteurs et des membres du groupe
d'étude ACTG A5095.
Contributions des auteurs: Dr Gulick a eu un accès complet
à toutes les données de l'étude et accepte la
responsabilité de l'intégrité des données et
de l'exactitude de l'analyse des données.
Conception et schéma de l'étude: Gulick,
Ribaudo, Shikuma, Acosta, Schouten, Bastow, Klingman, Kuritzkes.
Recueil des données: Gulick, Ribaudo, Squires, Pilcher,
Koletar, Carlson, Reichman, Bastow, Kuritzkes.
Analyse et interprétation des données: Gulick,
Ribaudo, Lalama, Schackman, Meyer, Schouten, Murphy, Klingman, Kuritzkes.
Rédaction du manuscrit: Gulick, Ribaudo, Lalama,
Kuritzkes.
Revue critique du manuscrit: Gulick, Ribaudo, Shikuma, Lalama,
Schackman, Meyer, Acosta, Schouten, Squires, Pilcher, Murphy, Koletar,
Carlson, Reichman, Bastow, Klingman, Kuritzkes.
Analyse statistique: Ribaudo, Lalama.
Aide administrative, technique, et matérielle: Schackman,
Meyer, Acosta, Squires, Murphy, Reichman, Bastow, Klingman.
Supervision de l'étude: Gulick, Shikuma, Schouten,
Pilcher, Murphy, Koletar, Carlson.
Liens financiers: le Dr Gulick déclare avoir reçu des
bourses de recherche (décernées à Cornell University) de
Boehringer-Ingelheim Gilead, et de Merck; il a travaillé comme ad hoc
consultant pour Boehringer-Ingelheim, Bristol-Myers Squibb, Gilead,
GlaxoSmithKline, et Merck et a reçu des honoraires en tant
qu'orateur de Bristol-Myers Squibb, Gilead, et Merck. Le Dr Shikuma
déclare avoir eu une affiliation ou une implication financière
avec Boehringer-Ingelheim, Bristol-Myers Squibb, Gilead, et GlaxoSmithKline.
Le Dr Acosta rapporte avoir travaillé comme consultant pour
Bristol-Myers Squibb, Merck, Monogram, Roche, et Tibotec. Le Dr Squires
déclare avoir reçu un soutien sous la forme de bourse de
recherche de Boehringer-Ingelheim, Bristol-Myers Squibb, Gilead Sciences,
Merck, Numoda, et Tibotec; il a siégé au bureau scientifique
consultatif de Boehringer-Ingelheim, Bristol-Myers Squibb, Gilead Sciences,
Incyte, Merck, Pfizer, et Tibotec; et a été orateur pour
Boehringer-Ingelheim, Bristol-Myers Squibb, Gilead Sciences, et
GlaxoSmithKline. Le Dr Pilcher déclare avoir reçu une bourse de
recherche de Bristol-Myers Squibb et de Gilead. Le Dr Murphy déclare
avoir été consultant et avoir reçu une bourse de
recherche de Bristol-Myers Squibb. Le Dr Carlson déclare avoir
reçu des bourses de recherche de Bristol-Myers Squibb et a servi de
consultant à Gilead. Le Dr Reichman déclare être
affilié à GlaxoSmithKline pour des royalties sur le vaccin human
papillomavirus. Le Dr Kuritzkes déclare être consultant et avoir
reçu des honoraires d'orateur pour Boehringer-Ingelheim,
Bristol-Myers Squibb, et GlaxoSmithKline; il a été consultant et
a reçu des bourses de recherche pour Bayer.
Aucun des auteurs ou du personnel clinique ou des laboratoires n'a
reçu de compensation directe des compagnies pharmaceutiques pour leur
participation à cette étude. Tous les auteurs et le personnel
clinique et des laboratoires ont reçu un soutien partiel du National
Institute of Allergy and Infectious Diseases (NIAID) par accord avec le AIDS
Clinical Trials Group. Les représentants pharmaceutiques du groupe du
protocole ont été compensés en tant que salariés
par leur compagnie pharmaceutique respective.
Financement/soutien: cette étude a
bénéficié du soutien sous la forme de bourses du NIAID,
National Institutes of Health (AI 38858 [AIDS Clinical Trials Group {ACTG}
Central Grant], AI 01781, AI 25859, AI 25868, AI 25879, AI 25897, AI 25903, AI
25915, AI 25924, AI 27658, AI 27659, AI, 27660, AI 27661, AI 27664, AI 27668,
AI 27670, AI 27673, AI 27675, AI 27767, AI 28697, AI 32775, AI 32782, AI
34832, AI 38855, AI 39156, AI 42848, AI 42851, AI 46339, AI 46370, AI 46376,
AI 46381, AI 46386, AI 50410, AI 51966, RR00044, RR00046, RR00047, RR00052,
RR00096, RR00865, RR02635, et de sous-contrats AI 38858 du Virology Support
Laboratories at Brigham and Women's Hospital, the University of Alabama,
the University of Colorado Health Sciences Center, the University of North
Carolina, et de la Vanderbilt University). Boehringer-Ingelheim, Bristol-Myers
Squibb, et GlaxoSmithKline ont tous fourni généreusement les
traitements à l'étude et Bristol-Myers Squibb ainsi que
GlaxoSmithKline ont également fourni un financement pour les tests HIV
RNA et métaboliques.
Rôle des sponsors: cette étude était
conçue et menée par les investigateurs ACTG. Les
représentants des compagnies pharmaceutiques dans le groupe du
protocole ont eu l'opportunité de commenter le schéma de
l'étude mais toutes les décisions finales sur le
schéma ont été faites par les investigateurs. Le NIAID a
donné son approbation finale à l'étude avant son
application. La conduite de l'étude a été
entièrement sous la responsabilité des investigateurs, avec un
contrôle réglementaire du NIAID. Le recueil des données,
leur contrôle et leur interprétation ont été
entièrement sous la responsabilité des investigateurs ACTG. Tous
les membres du groupe du protocole, y compris les représentants des
compagnies pharmaceutiques et du NIAID, ont eu l'opportunité de
commenter l'interprétation des données, mais les
décisions finales concernant l'interprétation des
données ont été la prérogative des investigateurs
ACTG. Le manuscrit a été préparé par une
équipe rédactionnelle comprenant les Drs Gulick, Ribaudo,
Lalama, et Kuritzkes et a circulé parmi les co-auteurs pour revue,
commentaires, et approbation. Tous les auteurs ayant donné leur
approbation au manuscrit, celui-ci a circulé auprès des membres
de l'équipe du protocole, y compris auprès des
représentants des compagnies pharmaceutiques et du NIAID, pour revue et
commentaires.
Le manuscrit a aussi été revu en interne par le ACTG
scientific leadership et par le ACTG Statistics and Data Analysis Center avant
sa soumission. La responsabilité finale pour approbation du manuscrit a
été dévolue aux auteurs.
Groupe d'étude ACTG A5095: Carol Bick, PhD (Indiana
University School of Medicine) and Alison Boyle, MT (University of California,
Los Angeles), study laboratory technologists; Valery Hughes, FNP (Weill
Medical College of Cornell University), study field representative; Anne
Kmack, BS, and Sandra Oyola, MT (Frontier Science and Technology Foundation),
study data managers; AnaMartinez, RPh (Division of AIDS, NIAID), study
pharmacist; Monica Murphy, MT, and Nancy Webb, MS (Frontier Science and
Technology Foundation), laboratory data coordinators; Vinny Parillo (ACTG
Community Constituency Group), community representative; Sally Snyder, BS
(Social & Scientific Systems Inc), study specialist; Doug Ferriman,
PharmD, Michael Imperiale, MD, Marita McDonough, MPA, RN, Jerry Stern, MD,
Stephen Storfer, MD (Boehringer-Ingelheim-Roxane Laboratories); Awny
Farajallah, MD, Michael Giordano, MD, Kelly Morrissey, RN, MSN, Jeffery Olson,
PharmD, Lynn Rugh, Kirk Ryan, PharmD, Michael Soccodato, Mary Swingle, RN,
Shulin Wang, MD (Bristol-Myers Squibb); Richard Fallis, BA, James Rooney, MD,
Stephen Smith (Gilead Sciences); Cindy Brothers, MSPH, Christina Hill-Zabala,
MD, Joe Mrus, MD, Keith Pappa, PharmD, Trevor Scott, PhD, Jerry Tolson, PhD,
and Amy Van Kempen, MEd (GlaxoSmithKline). Personnel des laboratoires:
Michelle Marcial, BS, Daniel Eggers, BS (Brigham and Women's Hospital);
Richard D'Aquila, MD, Lorraine Sutton, BA (Vanderbilt University);
Russell Young, MS (University of Colorado Health Sciences Center); Victoria
Johnson, MD, Darren Hazelwood, BS, Julia Parker, BA (University of Alabama at
Birmingham); Susan Fiscus, PhD, Leslie Petch, PhD, Tiffany Tribull, MS
(University of North Carolina, Chapel Hill). Membres des centres
participants: David Currin, RN, Lisa Danoit, RN, Kim Epperson, RN
(University of North Carolina); Donna McGregor, NP (Northwestern University);
Oluwatoyin Adeyemi, MD (Cook County Hospital); Harold Kessler, MD
(Rush-Presbyterian-St Luke's Medical Center); William Maher, MD, Diane
Gochnour, RN, Mark Hite, RN (Ohio State University); Ron Mitsuyasu, MD (UCLA
School of Medicine); Mallory Witt, MD, Mario Guerrero, MD (Harbor-UCLA Medical
Center); Carol Greisberger, RN, Roberto Corales, DO, Christine Hurley, RN
(University of Rochester Medical Center); SantiagoMarrero, MD, Olga Mendez,
MD, Irma Torres, RN, MSN (University of Puerto Rico); Julie Richardson,
PharmD, Janet Hernandez, RN, Scott Hamilton, RN (Indiana University Hospital);
Jose Castro, MD, Hector Bolivar, MD, Margaret Fischl, MD (University of
Miami); Deborah McMahon, MD, Sharon Riddler, MD (University of Pittsburgh);
Princy Kumar, MD (Georgetown University Medical Center); Sue Swindells, MD
(University of Nebraska); Jeffery Meier, MD (University of Iowa); Henry
Balfour, Jr, MD (University of Minnesota); Paula Potter, RN, BSN, Julie
Hoffman, RN, Francesca Torriani, MD (University of California, San Diego);
Martha Silberman, RN, Nathan Thielman, MD, Kenneth Shipp, RPh (Duke University
Medical Center); David Haas, MD, Janet Nicotera, RN, BSN, Jie Wang, RN
(Vanderbilt University); Mussolini Africano, PA-C, Luis Mendez, BS, Connie
Funk, RN, BSN (University of Southern California); Judith Feinberg, MD, Diane
Daria, BSN, Carol Colegate, RPh (University of Cincinnati); Tianna Petersen,
MS, Philip Keiser, MD (University of Texas, Southwestern Medical Center);
David Clifford, MD, Mark Rodriguez, RN, BSN, Kimberly Gray, RN, MSN Washington
University); James Scott, RN, BSN, Cathi Basler, RN, MSN, Steven Johnson, MD
(University of Colorado Health Sciences Center); Amy Sbrolla, RN, BSN, Neah
Kim, MSN, FNP, Jon Gothing, RN (Harvard University); Jeffery Lennox, MD (Emory
University); Kerry Upton, RN, BSN, J. Michael Kilby, MD (University of Alabama
at Birmingham); Todd Stroberg, RN (Cornell University); Jolene Noel-Connor,
RN, Madeline Torres, BSN (Columbia University); Janet Forcht, RN, Mary Volger,
MD (New York University); Timothy Flanigan, MD, Karen Tashima, MD, Helen
Sousa, LPN (The Miriam Hospital); Ann Conrad, RN, Jane Baum, BSN, RN (Case
Western Reserve University); Deb Him, RPh, Wayne Wagner, MSW, RN (University
of Pennsylvania); Ann Collier, MD, Becky Royer, PA-C, Laura Olin, ARNP
(University of Washington); Sandra Valle, PA-C, Sylvia Stoudt, RN, Deborah
Slamowitz, RN, BSN (Stanford University); Joanne Frederick, RN, Scott Souza,
PharmD, Debra Ogata-Arakaki, RN (University of Hawaii); Gwen Costantini, FNP,
Ronald D'Amico, MD, Donna Mildvan, MD (Beth Israel Medical Center);
Dorcas Baker, RN, Aruna Subramanian, MD (Johns Hopkins University);
WilliamO'Brien, MD, William Silkowski, RN, BSN University of Texas,
Galveston); Diane Havlir, MD, Jody Lawrence, MD, Joann Volinski, RN (San
Francisco General Hospital); Richard Pollard, MD, Melissa Schreiber, PA
(University of California, Davis). Analyse statistique: For this
National Institutes of Health – sponsored study of the ACTG, the
statistical analysis was conducted by Heather Ribaudo, PhD, and Christina
Lalama, MS, of the Statistical and Data Analysis Center at Harvard School of
Public Health, Boston, Mass. Présentation antérieure:
Présenté partiellement au 45th Annual Interscience
Conference on Antimicrobial Agents and Chemotherapy; December 16-19, 2005;
Washington, DC. Abstract H-416a.
Affiliations des auteurs: Weill Medical College of Cornell
University, New York, NY (Drs Gulick and Schackman); Statistical and Data
Analysis Center, Harvard School of Public Health, Boston, Mass (Dr Ribaudo and
Ms Lalama); University of Hawaii, Honolulu (Dr Shikuma); Quest Diagnostics
Inc, Baltimore, Md (Dr Meyer); University of Alabama, Birmingham (Dr Acosta);
University of Washington, Seattle (Dr Schouten); University of Southern
California Medical Center, Los Angeles (Dr Squires); University of North
Carolina, Chapel Hill (Dr Pilcher); Northwestern University, Chicago, Ill (Dr
Murphy); Ohio State University, Columbus (Dr Koletar); University of
California, Los Angeles (Dr Carlson); University of Rochester, Rochester, NY
(Dr Reichman); Social & Scientific Systems Inc, Silver Spring, Md (Ms
Bastow); Division of AIDS, National Institute of Allergy and Infectious
Disease, Bethesda, Md (Dr Klingman); and Brigham and Womens'Hospital and
Harvard Medical School, Boston (Dr Kuritzkes). Dr Squires est actuellement
affilié au Jefferson Medical College, Philadelphia, Pa.
BIBLIOGRAPHIE
| |
1. Panel on Clinical Practices for the Treatment of Infection.Guidelines for the use of antiretroviral agents in HIV-infected adultsand adolescents. May 4, 2006.http://www.aidsinfo.org.Accessibility verified 12, 2006.
2. Yeni PG, Hammer SM, Hirsch M, et al. Treatment for adult HIVinfection: 2004 recommendations of International AIDS Society – USAPanel. JAMA. 2004;292: 251-265.
FREE FULL TEXT
3. Gulick RM, Mellors JW, Havlir D, et al. Treatment with indinavir,zidovudine, and lamivudine in adults with human immunodeficiency virusinfection and antiretroviral therapy. N Engl J Med.1997; 337:739.
4. Hammer SM, Squires KE, Hughes MD, et al. A controlled trial of twonucleoside analogues plus indinavir in persons with human immunodeficiencyinfection and CD4 cell counts of 200 per cubic millimeter or less.N Engl J Med. 1997;337: 725-733.
PUBMED
5. Palella FJ Jr, Delaney KM, Moorman AC, et al. Declining morbidityand mortality among patients advanced human immunodeficiency virus infection.N Engl J Med. 1998;338: 853-860.
PUBMED
6. Mouton Y, Alfandari S, Valette M, et al. Impact protease inhibitorson AIDS-defining events and hospitalizations in 10 French AIDS referencecentres. AIDS. 1997;11: F101-F105.
PUBMED
7. Dornadula G, Zhang H, VanUitert B, et al. Residual HIV-1 RNA inblood plasma of patients taking suppressive highly active antiretroviraltherapy. JAMA. 1999;282: 1627-1632.
FREE FULL TEXT
8. Furtado MR, Callaway DS, Phair JP, et al. Persistence of HIV-1transcription in peripheral-blood mononuclear cells in patients receivingpotent antiretroviral therapy. N Engl J Med.1999; 340:1614-1622.
PUBMED
9. Weverling GJ, Lange JMA, Jurriaans S, et al. Alternative multidrugregimen provides improved suppression of HIV-1 replication over tripletherapy. AIDS. 1998;12: F117-F122.
PUBMED
10. van Pragg RM, Wit FWNM, Jurriaans S, de Wolf F, Prins JM, LangeJMA. Improved long-term suppression of HIV-1 replication with a triple-classmultidrug regimen compared with standard of care antiretroviral therapy.AIDS. 2002; 16:719-725.
PUBMED
11. Markowitz M, Louie M, Hurley A, et al. A novel antiviralintervention results in more accurate assessment of human immunodeficiencyvirus type 1 replication dynamics and T-cell decay in vivo. JVirol. 2003; 77:5037-5038.
FREE FULL TEXT
12. Louie M, Hogan C, Di Mascio M, et al. Determining the relativeefficacy of highly active antiretroviral therapy. J InfectDis. 2003; 187:896-900.
FREE FULL TEXT
13. Ramratnam B, Ribeiro R, He T, et al. Intensification ofantiretroviral therapy accelerates the decay of the HIV-1 latent reservoir anddecreases, but does not eliminate, ongoing virus replication. JAcquir Immune Defic Syndr. 2004;35: 33-37.
14. Fischl MA, Ribaudo HJ, Collier AC, et al. A randomized trial of 2different 4-drug antiretroviral regimens versus a 3-drug regimen in advancedhuman immunodeficiency virus disease. J Infect Dis.2003; 188:625-634.
FREE FULL TEXT
15. Shafer RW, Smeaton LM, Robbins GK, et al. Comparison of four-drugregimens and pairs of sequential three-drug regimens as initial therapy forHIV-1 infection. N Engl J Med. 2003;349: 2304-2315.
PUBMED
16. Yeni P, Cooper DA, Aboulker JP, et al; INITIO Trial InternationalCo-ordinating Committee. Virological and immunological outcomes at 3 yearsafter starting antiretroviral therapy with regimens containing nonnucleosidereverse transcriptase inhibitor, protease inhibitor, or both in INITIO:open-label randomised trial. Lancet.2006; 368:287-298.
PUBMED
17. Gulick RM, Ribaudo HJ, Shikuma CM, et al. Triplenucleoside regimensvs. efavirenz-containing regimens for the initial treatment of HIV-1infection: AIDS Clinical Trials Group (ACTG) Study A5095. N Engl JMed. 2004; 350:1850-1861.
PUBMED
18. Chesney MA, Ickovics JR, Chambers DB, et al. Selfreported adherenceto antiretroviral medications among participants in HIV clinical trials: theAACTG adherence instruments. AIDS Care.2000; 12:255-266.
PUBMED
19. Regulatory Compliance Center. Division of AIDS table forgrading severity of adult adverse experiences. August1992.http://rcc.tech-res-intl.com/tox_tables.htm.Accessibility verified June 12, 2006.20. Liang K-Y, Zeger SL. Longitudinal data analysis using generalizedlinear models. Biometrika. 1986;73: 13-22.
FREE FULL TEXT
21. Grambsch P, Therneau T. Proportional hazards tests and diagnosticsbased on weighted residuals. Biometrika.1994; 81:515-526.
FREE FULL TEXT
22. Clavel F, Hance AJ. HIV drug resistance. N Engl JMed. 2004; 350:1023-1035.
PUBMED
23. Staszewski S, Morales-Ramirez J, Tashima KT, et al. Efavirenz pluszidovudine and lamivudine, efavirenz plus indinavir, and indinavir pluszidovudine and lamivudine in the treatment of HIV-1 infection in adults.N Engl J Med. 1999;341: 1865-1873.
PUBMED
24. Gallant JE, Staszewski S, Pozniak AL, et al; 903 Study Group.Efficacy and safety of tenofovir DF vs stavudine in combination therapy inantiretroviralnaive patients: a 3-year randomized trial.JAMA. 2004; 292:191-201.
FREE FULL TEXT
25. Albrecht MA, Bosch RJ, Hammer SM, et al. Nelfinavir, efavirenz, orboth after the failure of nucleoside treatment of HIV infection. NEngl J Med. 2001; 345:398-407.
PUBMED
26. Lupo L, Maa J-F, Dezii C, Said D, Bessen L, Hodder S. Efficacyof efavirenz in different racial groups. In: Abstracts of theSixth International Congress on Drug Therapy in HIV Infection;November 17-21, 2002; Glasgow, Scotland. AbstractP61.27. Robbins GK, De Gruttola V, Shafer RW, et al. Comparison ofsequential three-drug regimens as initial therapy for HIV-1 infection.N Engl J Med. 2003;349: 2293-2303.
PUBMED
28. Wegner S, Vahey M, Dolan M, et al. Racial differences inclinical efficacy of efavirenz-based antiretroviral therapy. In:Abstracts of the 9th Conference on Retroviruses and OpportunisticInfections; February 24-28, 2002; Seattle, Wash. Abstract428-W.29. Moore R, Keruly J, Gebo K, Lucas G. Racial differences inefavirenz discontinuation in clinical practice. In: Abstracts ofthe 12th Conference on Retroviruses and Opportunistic Infections;February 25, 2005; Boston, Mass. Abstract 619.30. Haddy TB, Rana SR, Castro O. Benign ethnic neutropenia: what is anormal absolute neutrophil count? J Lab Clin Med.1999; 133:15-22.
PUBMED
31. Haas DW, Ribaudo HJ, Kim RB, et al. Pharmacogenetics of efavirenzand central nervous system side effects: an Adult AIDS Clinical Trials Groupstudy. AIDS. 2004;18: 2391-2400.
PUBMED
32. World Health Organization. Scaling up antiretroviraltherapy in resource-limited settings: treatment guidelines for a public healthapproach. 2003, updated 2005.http://www.who.int/3by5/publications/documents/arv_guidelinesy/en/.Accessibility verified June 12, 2006.33. Barreiro P, Soriano V, Casas E, Gonzalez-Lahoz J. Different degreeof immune recovery using antiretroviral regimens with protease inhibitors ornon-nucleosides. AIDS. 2002;16: 245-249.
PUBMED
34. Manfredi R, Calza L, Chiodo F. First-line efavirenz versuslopinavir-ritonavir-based highly active antiretroviral therapy for naivepatients. AIDS. 2004;18: 2331-2333.
PUBMED
35. Torti C, Maggiolo F, Patroni A, et al; MASTER Cohort. Exploratoryanalysis for the evaluation of lopinavir/ritonavir- versus efavirenz-basedHAART regimens in antiretroviral-naive HIV-positive patients: results from theItalian MASTER Cohort. J Antimicrob Chemother.2005; 56:190-195.
FREE FULL TEXT
36. Hicks C, King MS, Gulick RM, et al. Long-term safety and durableantiretroviral activity of lopinavir/ritonavir in treatment-naive patients: 4year follow-up study. AIDS. 2004;18: 775-779.
PUBMED
37. Hirsch MS, Brun-Vezinet F, Clotet B, et al. Antiretroviral drugresistance testing in adults infected with human immunodeficiency virus type1: 2003 recommendations of an International AIDS Society – USA Panel.Clin Infect Dis. 2003;37: 113-128.
FREE FULL TEXT
38. Bossi P, Colin D, Bricaire F, Caumes E. Hypersensitivity syndromeassociated with efavirenz therapy. Clin Infect Dis.2000; 30:227-228.
FREE FULL TEXT
39. Clay PG. The abacavir hypersensitivity reaction: a review.Clin Ther. 2002;24: 1502-1514.
PUBMED
40. Mallal S, Nolan D, Witt C, et al. Association between presence ofHLA-B*5701, HLA-DR7, and HLA-DQ3 and hypersensitivity to HIV-1reversetranscriptase inhibitor abacavir. Lancet.2002; 359:727-732.
PUBMED
41. Phillips EJ, Wong GA, Kaul R, et al. Clinical and immunogeneticcorrelates of abacavir hypersensitivity. AIDS.2005; 19:979-981.
PUBMED
ARTICLE EN RAPPORT
Comparaison de schémas thérapeutiques comportant 3 ou 4 antirétroviraux pour le traitement initial de l'infection par le VIH 1: Un essai contrôlé randomisé
Roy M. Gulick, Heather J. Ribaudo, Cecilia M. Shikuma, Christina Lalama, Bruce R. Schackman, William A. Meyer, III, Edward P. Acosta, Jeffrey Schouten, Kathleen E. Squires, Christopher D. Pilcher, Robert L. Murphy, Susan L. Koletar, Margrit Carlson, Richard C. Reichman, Barbara Bastow, Karin L. Klingman, Daniel R. Kuritzkes, et pour le groupe d'étude AIDS Clinical Trials Group (ACTG) A5095
JAMA. 2006;296:769-781.
Résumé
| Texte Complet
|