How It’s Made: The Synapse
Abstract
How are synapses made? This question is one of the most important issues in neurobiology today and has been the subject of intense study in recent years. This review focuses on the mechanisms of presynaptic terminal formation in the mammalian central nervous system. Building a synapse requires stabilization of contacts between axons and dendrites and formation of synaptic subcellular structures. Here, we discuss what determines where and when synapses form, how components of the nascent presynaptic terminal accumulate at the site of synapse formation, and whether assembly occurs via an ordered process dependent on a master organizer. Understanding synapse formation in the central nervous system is relevant for understanding and treating brain diseases as diverse as autism, epilepsy, anxiety disorders, brain injury, and Alzheimer’s disease.
Introduction
The neural circuits that govern perception and behavior are composed of networks of neurons that communicate with one another via synapses. As our brains develop and mature, the roughly 10–20 billion neurons that comprise the human cerebral cortex are presented with the enormous and complex task of forming trillions of synapses. How this is accomplished is not yet clear, but the mechanisms of synapses formation between neurons of the mammalian brain have been intensely studied in recent years.
A synapse has two major components: the presynaptic terminal of the signaling neuron and the postsynaptic density of the receiving neuron (Figure 1A). This review will focus mainly on the formation of presynaptic terminals. These terminals contain clusters of small, uniformly-sized synaptic vesicles and an electron-dense region at the plasma membrane called the active zone. In response to electrical activity in the presynaptic terminal, synaptic vesicles fuse with the active zone membrane. Upon fusion, chemical neurotransmitters are released from synaptic vesicles. Secreted transmitter diffuses across the synaptic cleft and binds to neurotransmitter receptors in the postsynaptic membrane to activate signaling in the postsynaptic neuron. At each stage, specialized protein complexes are required for this highly regulated signaling between neurons.
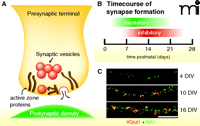
Synapse structure and time course of synapse formation. A. Excitatory synapses are composed of a presynaptic terminal positioned opposite a postsynaptic density. The presynaptic terminal contains clusters of neurotransmitter-containing synaptic vesicles and a dense meshwork of proteins that make up the active zone and its matrix. B. In the rodent cortex, synapses are formed during a protracted period. Excitatory synaptogenesis begins during the first postnatal week and peaks during the second week. The bulk of inhibitory synapse formation is delayed relative to excitatory synaptogenesis. Eyes and ear canals open near the end of the second postnatal week. C. Images of cortical excitatory synapses in vitro at 4, 10, and 16 DIV. Images show overlays of VGlut1 (red) and NR1 (green) immunostaining. Scale bar, 10 μm. (DIV, days in vitro.)
In the television series “How It’s Made,” the viewer watches as objects ranging from marbles to bicycles to carburetors are made on assembly lines. For complex objects, a single component is added, one at a time, until the product is completely assembled. One assumes that most things are made in an orderly, step-by-step manner. When examining a synapse, it would be logical to speculate that an analogous process takes place—that a synapse is made up of many individual “pieces” that interact with each other to become one functional unit. As discussed below, however, recent research has rendered this simple model incomplete at best.
Intitiation of Synapse Formation: When and Where Do Synapses Form?
In the rodent cortex and hippocampus, the bulk of synaptogenesis occurs over a protracted period during the first few postnatal weeks (Figure 1B). Excitatory synapse formation occurs very rapidly during the second postnatal week, while inhibitory synapse formation is delayed, peaking during the end of the second week and into the third week (Figure 1B). In vitro, the time course of synapse formation is similar to that observed in vivo (Figure 1C). Importantly, formation of individual synapses is highly unsynchronized, even for a given neuron, and an individual synapse can be assembled over a time-scale of tens of minutes to a few hours (1–5).
The majority of synapses in the cortex and hippocampus are axo-dendritic synapses, formed at sites of contact between axons and dendrites. Axo-axonic synapses also occur (e.g., between GABAergic chandelier neurons and the axon initial segments of excitatory neurons), but are present at a much lower frequency (6, 7). Most of the synapses made between neurons of the central nervous system (CNS) are formed en passant, meaning they occur along the length of the axon rather than only at the far axon termination. Therefore, in principle, synapse formation could be induced by axonal growth cones contacting dendrites, dendritic growth cones contacting axons, or dendritic shaft filopodia contacting nearby axons or vice versa (Figure 2).
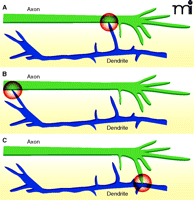
Different forms of synaptogenesis. Synapse formation is often induced by contact between dendritic filopodia and axons (A), but can also be initiated by contact between dendritic growth cones and axons (B) or axonal growth cones and dendrites (C).
Imaging studies in vitro and in vivo have demonstrated that excitatory synapse formation is often initiated by contact between dendritic filopodia and axons (8–11). Most contacts between dendritic filopodia and axons are not stabilized; therefore, unidentified factors from or within axons probably determine where axo-dendritic contacts stabilize and synapses form. In support of this hypothesis, axo-dendritic contacts between cortical neurons stabilize preferentially at sites that are intrinsic within cortical axons and defined prior to contact formation (5). It will be important to determine which molecules comprise these sites. Sec6, a component of the exocyst complex, can be found at these sites; however, whether the exocyst participates in synapse formation is not known. Although axonal adhesion proteins are present at these sites, no one axonal adhesion protein is present at all sites (S. Sabo, unpublished observations). This finding could mean that adhesion proteins: 1) do not define these sites; 2) are functionally redundant at sites of synapse formation; or 3) define sites that will become synapses that are qualitatively different from one another.
Once axo-dendritic contact is stabilized, the induction of synapse formation may be mediated by a set of transmembrane proteins that interact across the eventual synaptic cleft. Several such trans-synaptic proteins have been identified in the past decade (Figure 3). Some of these trans-synaptic interactions are mediated by heterophilic adhesion. For example, the interaction of neuroligins on dendrites with neurexins on axons is sufficient to induce assembly of both excitatory and inhibitory presynaptic terminals within axons (12–15). Leucine rich–repeat transmembrane neuronal proteins (LRRTMs) located at dendrites can induce presynaptic terminal formation through trans-synaptic interaction with neurexin at the surface of axons, as well (16–19). The receptor EphB2 can induce presynaptic development by binding the ligands ephrin-B1 and -B2 in axons (20, 21). Similarly, the interaction of dendritic netrin-G ligand-3 (NGL-3) with leukocyte common antigen-related (LAR) protein tyrosine phosphatase (PTP) or other PTPs in axons can induce presynaptic assembly (22–24). The interaction of the proteins neph and nephrin may also possess synapse-inducing activity because their C. elegans orthologs, syg-1 and syg-2, determine where synapses form in C. elegans hermaphrodite-specific neurons (HSN) (25–27). Homophilic trans-synaptic adhesion interactions can also induce synapse assembly. Both synCAM (15, 28) and amyloid precursor protein (APP) (29) are capable of inducing presynaptic terminal assembly via homophilic interactions. In fact, these interactions may not be truly homophilic because synCAM1 might preferentially interact with synCAM2 (30), and APP might also interact with amyloid precursor-like protein 2 (APLP2) in this context (29).
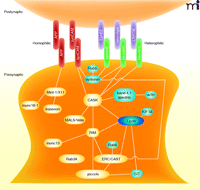
Schematic of synaptogenic trans-synaptic adhesion molecules and their key downstream interactions. Homophilic trans-synaptic interactors are shown in pink. Heterophilic trans-synaptic synapse inducers are shown in light purple (postsynaptic) and green (presynaptic). Within the presynaptic terminal, demonstrated protein-protein interactions are shown with solid lines. For proteins highlighted in yellow, knockout mice or knockdown in mammals exhibit either no synaptogenesis phenotype or only mild changes in synapse structure. α-Liprin is highlighted in dark blue because, although it has been shown to be important for synapse formation in C. elegans and Drosophila (106–110), it has not yet been tested for presynaptic terminal formation in mammals. RIM, Rab3-Interacting Molecule; GIT, G protein–coupled receptor kinase-interactor-1; CASK, calcium/calmodulin-dependent serine kinase.
Intriguingly, the formation of presynaptic terminals can be induced through contact between axons and non-neuronal cells (e.g., HEK-293 cells or cos-7 cells) that express neuroligin, synCAM, LRRTM, EphB2, NGL, or APP (12, 16–22, 28, 29). The induced presynaptic terminals appear to be functional and competent for neurotransmitter release (28, 31). Moreover, the newly formed presynaptic terminals are morphologically indistinguishable from normal presynaptic terminals, with clusters of small, clear synaptic vesicles located adjacent to an active zone at the site of contact (12). Finally, these terminals express the molecular markers that are representative of presynaptic terminals.
Not all synaptic adhesion molecules are sufficient to induce synapse formation. In the hemi-synapse induction assay described above, neither N-cadherin, NCAM-140, TAG-1, nor L1 could induce presynaptic terminal formation (12, 15). Intriguingly, although N-cadherin cannot itself induce presynaptic terminal formation, N-cadherin cooperates with neuroligin in the postsynaptic neuron to control the induction of presynaptic terminals (32).
How trans-synaptic adhesion is translated within the axon to recruit synaptic proteins is not understood. However, the presynaptic terminals induced by these trans-synaptic adhesion proteins are indistinguishable from one another, suggesting their effects are mediated by a common downstream signal. Consistent with this idea, swapping of cytoplasmic domains of APP and synCAM still results in trans-synaptic adhesion capable of inducing presynaptic terminal formation (29).
Transport of Synaptic Proteins to Sites of Contact
Synapse assembly requires delivery of all of the specialized active zone and synaptic vesicle proteins to the right place at the right time. Instead of being individually transported, many synaptic proteins are packaged together in the cell body and transported in larger aggregates to a developing synapse. In particular, two types of vesicular organelles, each containing specific cargo, are currently thought to participate in synaptogenesis: 1) synaptic vesicle protein transport vesicles (STVs), and 2) piccolo-bassoon transport vesicles (PTVs) (Figure 4).
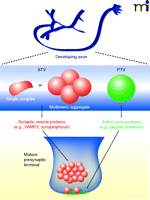
In developing axons, synaptic vesicle and active zone proteins are delivered to developing presynaptic terminals via distinct precursor organelles. Synaptic vesicle protein transport vesicles (STVs) (red) carry a variety of synaptic vesicle proteins and have morphologies ranging from tubulovesicular structures to clusters of small clear vesicles that resemble synaptic vesicles. Piccolo-bassoon transport vesicles (PTVs) (green) carry several active zone proteins and have been described as dense-core vesicles. It is not clear which cues recruit these transport intermediates to developing synapses.
Active zone proteins are transported to nascent synapses in PTVs. Isolated PTVs were first described as dense-core vesicles of approximately 80nm in size (33). More recently, however, evidence suggests PTVs might exist as aggregates of dense and clear-core vesicles, possibly connected by undefined electron-dense material (34). Active zone proteins and exocytic regulators that are associated with PTVs include piccolo, bassoon, N-cadherin, syntaxin, synaptosomal-associated protein 25 kDa (SNAP25), Rab3a, Rab3c, chromogranin B, Rab3-interacting molecule (RIM), Munc13, and Munc18 (4, 33). These proteins are assembled into PTVs through a process that at least partially depends on the trans-Golgi network. Bassoon must be post-translationally processed via the trans-Golgi network before being incorporated into PTVs, and PTVs containing bassoon and piccolo accumulate around (although not within) the Golgi apparatus in hippocampal neurons (34, 35).
Proteins associated with synaptic vesicles are thought to be transported to nascent synapses via STVs, which consist of a variety of structures, including tubulovesicular and clear-core vesicles (1, 36, 37). These structures originate from the trans-Golgi region and are able to undergo cycling throughout the axon (5, 36–40). Proteins transported by STVs include SV2, synapsin Ia, amphiphysin I, the calcium channel subunit α1a, vesicle-associated membrane protein 2 (VAMP2), synaptophysin, and synaptotagmin (1, 36, 37).
Once assembled in the cell body, PTVs and STVs are specifically transported to axons through a variety of mechanisms (41, 42). Upon entering the axon, PTVs and STVs show similar characteristics of movement. Both types of transport vesicles display movement in both the retrograde and anterograde directions (1, 4, 36, 37, 43). When moving, STVs can reach speeds ranging from 0.1–1.0 μm/sec, whereas PTVs move at a rate of 0.1–0.35 μm/sec (4, 36, 37, 43, 44). Both PTVs and STVs display saltatory movement. The frequent pauses in between bursts of movement may be important in synaptogenesis because sites where STVs repeatedly pause are more likely to develop synapses (5).
In axons, microtubules are arranged with their “plus-end” oriented distal and their minus-end proximal to the cell body. This alignment has implications for the transport of PTVs and STVs as each can move in both directions in the axon. A variety of molecular motors are responsible for this movement. For anterograde transport, PTVs and STVs associate with different kinesin superfamily motors (KIFs). KIFs are plus end–directed microtubule-based motors involved in numerous processes throughout the cell (45).
The kinesin-1 motor KIF5B is the main molecular motor for PTVs. Correct attachment of the motor to its cargo is needed for proper distribution of PTVs in the axon and for normal synapse development and function (44, 46). KIF5B has also been implicated in a number of other functions, including the axonal transport of mitochondria (47, 48). STVs, on the other hand, are transported by two different kinesin-3 motors. KIF1A was first associated with STVs when a KIF1A ortholog, unc-104, was found necessary for the correct localization of synaptic vesicles in C. elegans (49, 50). Likewise, mammalian KIF1A is required for proper transport of synaptic vesicle precursor proteins out of the cell body (51, 52). Later, a second motor, KIF1Bβ was implicated in the transport of synaptic vesicle proteins. KIF1Bβhas a similar cargo domain to that of KIF1A, which enables the two motors to bind similar targets. KIF1Bβ co-localizes with various synaptic vesicle proteins and transports STVs throughout the axon (53). Confirming its importance, KIF1Bβ knockout mice are defective in the axonal transport of synaptic vesicles (54).
Unlike anterograde transport, retrograde transport of STVs and PTVs occurs only through dynein, a minus end–directed microtubule-based motor protein (55–58). Disruption of the dynein motor complex results in an overall decrease in membranous organelle transport, and, in C. elegans, results in misaccumulation of both active zone proteins and synaptic vesicle proteins in distal portions of the neuron (59, 60). Likewise, distribution of active zone proteins is disrupted when PTV transport via dynein is reduced in mammals (61).
Rather than attaching directly to the motors that carry them, STVs and PTVs use linking molecules to bind to the appropriate motor. This combination of adaptors and motors might be one way that neurons ensure the correct transport of pre-synaptic proteins in the axon (62). As there are different motors that associate with STVs and PTVs, so there are different adaptors that link these cargos with their motors. Syntabulin links PTVs with the KIF5B motor, and the loss of this linkage reduces PTV distribution in axons and results in overall impaired synaptic function (44, 46, 48). For STVs, the specific molecules connecting them with the KIF1A and KIF1Bβ motors are less clear. Liprin-α has been implicated in linking STVs with KIF1A, whereas the Rab3 GDP-GTP exchange factor termed differentially expressed in normal versus neoplastic (DENN)/mitogen-activated protein kinase-activating death domain (MADD) may link vesicles containing the synaptic vesicle protein Rab3 with both KIF1A and KIF1Bβ (63–65). Although progress has been made, more work is needed to determine the identity and importance of linking molecules between STVs, PTVs, and their anterograde motors.
As both STVs and PTVs utilize dynein as their retrograde motor, it is likely the molecules that link the cargos to the motor are the same. The large multi-subunit protein dynactin has been well characterized as an essential part of most dynein-mediated motor activity (55, 66). With regard to STVs and PTVs, interruptions in the dynein-dynactin complex results in reduced transport of membranous organelles in squid axoplasm (59). Disrupting this complex also leads to an overall decrease in retrograde transport in a mouse model of amyotrophic lateral sclerosis and to aberrant distal accumulation of active zone proteins and synaptic vesicle–associated proteins in C. elegans (60, 67). This finding does not rule out the possibility of other linking molecules between dynein and STVs or PTVs. For example, the PTV marker protein bassoon directly interacts with dynein light chain and disrupting this interaction causes defects in PTV distribution in the axon (61). Again, more work needs to be done to fully elucidate the relationship between STVs, PTVs, and dynein.
How Does Synaptogenic Adhesion Induce Presynaptic Terminal Assembly?
What are the signals that link synaptic contact and trans-synaptic adhesion to trafficking and recruitment of synaptic vesicle and active zone precursors to the right place at the right time? Because many active zone proteins participate in multiple protein-protein interactions (Figure 3), the prevailing theory is that trans-synaptic adhesion induces an interaction with some scaffold-like active zone protein. The scaffold protein then recruits additional active zone proteins and eventually synaptic vesicle proteins through a chain or network of protein-protein interactions.
Calcium/Calmodulin-Dependent Serine Kinase (CASK)
Calcium/calmodulin-dependent serine kinase (CASK) stands out as perhaps the most interesting interaction partner because it can interact directly with synCAM and neurexin and indirectly with APP, via Mint-1/X11alpha (Figure 3). Recent experiments, however, have examined CASK knockout mice and neurons treated with RNAi to knockdown CASK expression (68). Eliminating or dramatically reducing CASK expression did not prevent synapse formation, supporting the argument that CASK is not the key synaptic organizer molecule downstream of axo-dendritic contact. Expression of CASK-interacting proteins, including neurexins, Mals/Velis, and Mints were decreased, but synaptic architecture was normal. Consistent with this idea, knockout of the CASK interacting proteins Mint-1 (69) or MALS/Velis (70) does not disrupt synapse formation, although synapse ultrastructure was not examined in these studies.
Syntenin
Syntenin interacts with synCAM, ephrin-B1, ephrin-B2, and neurexin (Figure 3) (21, 28), and induction of presynaptic terminal formation by EphB2–ephrin-B interactions is blocked by either expression of a dominant-negative syntenin mutant or of syntenin shRNAs (21). It remains unclear whether syntenin can also mediate the effects of other synapse inducers or whether elimination of syntenin prevents synapse formation in vivo. Aside from its interactions with synaptogenic adhesion proteins, syntenin interacts with the endocytic protein Rab5 and the active zone protein ERC/CAST (71). Through ERC/CAST, syntenin is indirectly linked to Rab6 (a small GTP-binding protein involved in membrane trafficking at the Golgi), liprin, piccolo, and RIM (72–75). ERC/CAST deletion does not alter the structure of excitatory presynaptic terminals, except for a small decrease in synaptic vesicle number (76).
Rab3-Interacting Molecule (RIM)
RIM interacts with multiple active zone proteins and with synaptic vesicle proteins (Figure 3). RIM directly interacts with the active zone proteins ERC/CAST and piccolo (73, 77). RIMs also interact with Rab3A, calcium channels, liprins, munc13-1, synaptotagmin, and RIM binding protein (RIMBP) (78). Elimination of RIM1α expression appears to decrease neurotransmitter release (78) and decrease the percentage of synaptic vesicles residing in the readily releasable pool (79), but synapse structure remains normal (78). These data suggest that RIM is not the key common factor mediating the induction of synapse assembly by trans-synaptic adhesion. In further support of this conclusion, RIMs have been proposed to be linked to synaptic vesicles both physically and functionally via Rab3A, but no changes in synapse morphology or function are observed upon knockout of Rab3-GEF, its guanine nucleotide exchange factor, or rabphilin (80–82).
Piccolo and Bassoon
Neither piccolo nor bassoon is required for presynaptic terminal assembly. Knockout or knockdown of piccolo does not cause any major changes in presynaptic terminal structure (83, 84). Similarly, bassoon knockouts do not exhibit morphological changes, although a fraction of synapses were non-functional (85). Because of their structural similarities, bassoon and piccolo might be functionally redundant during synapse formation. However, when bassoon is knocked down in piccolo knockout neurons, no electrophysiological changes or major structural changes to the active zone are observed at excitatory or inhibitory synapses, but a decrease in synaptic vesicle clustering is observed (83).
Intriguingly, none of the proteins tested so far––whether active zone proteins or synaptic vesicle proteins, and including many not discussed in depth here––are required for mammalian CNS synapse formation, even though many of them are important for presynaptic terminal function. This leads one to question whether any synaptic protein is required for synapse formation.
Is there no master organizer of presynaptic terminals that mediates CNS synapse assembly through protein-protein interactions with all synaptogenic adhesion molecules? Instead, might synaptogenic adhesion induce activation of a signal transduction pathway or pathways, promoting multiple protein interactions that are functionally redundant for synapse formation? Alternatively, an induced signal transduction pathway could act by stopping the transport of synaptic proteins at sites of synapse formation, and the synaptic machinery could essentially self-assemble. Such signals remain to be identified, but this will be an important next step for the field.
There is some evidence to support the idea that synaptic complexes can self-assemble. First, the presynaptic cytomatrix has previously been suggested to self-assemble (54). Remarkably, when active zones are biochemically isolated and disassembled by raising the pH in the presence of detergent, slow lowering of the pH results in reconstitution of the active zone matrix with normal structural features. Second, contact between an axon and poly-lysine-coated beads can bypass the requirement for synaptogenic adhesion proteins and induce self-assembly of presynaptic terminals (86–89). Third, a recent study using genetics to identify mutants defective for appropriate presynaptic terminal formation in C. elegans shows that mutations in the Arf-like small G protein Arl-8 cause presynaptic proteins to accumulate in proximal regions of axons (90). Electron microscopic analysis of the sites of ectopic localization reveals that small synaptic vesicles and electron-dense active zone–like structures appeared. These observations are consistent with the hypothesis that an accumulation of presynaptic proteins can self-assemble.
Recent observations are also consistent with the hypothesis that signals induced by synaptogenic adhesion could recruit synaptic proteins primarily through stopping of STV and PTV transport at sites of axo-dendritic contact. Contacts are specifically stabilized and synapses are preferentially formed at restricted sites within the axon, and these sites are identified as spots where STVs tend to stop their transport (5). Localized signals induced by trans-synaptic adhesion could inhibit either motor activity or interactions between cargo, adaptors, and/or motors. It will be important to test this hypothesis further in the future.
Disorders Related to Cortical Synapse and Circuit Development
Understanding synapse formation is clinically relevant because abnormal circuit development has been linked to disorders as diverse as autism, mental retardation, amblyopia, epilepsy, anxiety, depression, and schizophrenia (91–94). In addition to its obvious role during brain development, synapse formation continues to be important during adulthood. A recent report suggests that at least some anti-depressant actions are mediated by induction of synaptogenesis (94). Synaptogenesis has also been implicated as a form of plasticity [see (95)]. In addition, synapse formation is important for recovery after brain injury. Finally, the ability to promote synaptogenesis will be important for treatment of neurodegenerative diseases, such as Alzheimer’s disease, where synapse loss occurs prior to diagnosis. It will be important to determine whether the mechanisms of synapse assembly are the same in developing and mature systems.
Conclusions
Synapse formation occurs through a complex mixture of interactions and processes. Key aspects of what we have learned in recent years and discussed here about the process of constructing pre-synaptic terminals are summarized in the model shown in Figure 5. At most, only some of these interactions seem to occur in an orderly fashion, like the assembly lines mentioned above. More studies, extending the work described above, using techniques such as live imaging that capture the dynamics of the assembly process are necessary to reveal how synapses are made.
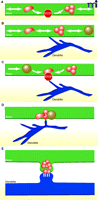
A model of synapse assembly. A. Before and during synapse formation, groups of synaptic vesicle and active zone proteins are transported along axons in precursor organelles, referred to as STVs (red) and PTVs (grey filled), respectively. STVs pause frequently at future sites of synapse formation. B. Axons and dendrites interact with one another. C. When axo-dendritic interactions occur at STV pause sites and trans-synaptic adhesion is engaged, contacts are stabilized. D. Synaptic proteins are deposited at the site of contact. E. Synaptic structures form, and synaptic proteins are sorted to their appropriate locations as the contact site differentiates into a functional synapse.
This review has focused on formation of synapses between neurons of the mammalian brain. It is important to note that there have been many excellent studies in recent years that have focused on neuromuscular synapse formation or used genetics in invertebrate models to identify molecular mechanisms of synapse formation. Although a comprehensive discussion of this literature is beyond the scope of this review, it will be exciting in the future to learn whether the molecular mechanisms of synapse formation are evolutionarily conserved. As an example, the PHR proteins (for PAM, highwire, rpm-1), serve an evolutionarily conserved function in neuromuscular synapse development, but their role in synapse formation in the mammalian brain is not yet known (96–105).
Acknowledgments
We apologize to the many authors whose work was not discussed owing to space constraints. We thank Michael Sceniak for helpful discussions. This work was supported by funds from the PhRMA Foundation.
- Copyright © 2010
References

Luke Bury, BS, joined the Sabo laboratory in the Department of Pharmacology at Case Western Reserve University in 2009 after graduating from Clemson University with a BS degree in biological science. He is currently investigating axonal trafficking of pre-synaptic proteins and how this affects the development of synapses.

Shasta L. Sabo, PhD, is Assistant Professor in the Departments of Pharmacology and Neurosciences at Case Western Reserve University. She received her doctoral degree from Rockefeller University in 2000. Her research focuses on the cellular and molecular mechanisms of synapse formation in the postnatal cerebral cortex. Send correspondence to SLS: E-mail shasta.sabo{at}case.edu; fax 216-368-5683.

