Regulation of ABC Transporters at the Blood-Brain Barrier: New Targets for CNS Therapy
Abstract
Worldwide, more than one billion people are affected by CNS disorders. Despite the huge demand for treatments, existing drugs have limited or no efficacy for some neurological diseases, including brain cancer and certain epilepsies. Furthermore, no effective therapies are available at all for some common disorders of the central nervous system (CNS) such as Alzheimer’s disease. ATP-binding cassette (ABC) transporters at the blood-brain barrier (BBB) have become increasingly important in the treatment and pathogenesis of CNS disorders. Here we highlight a novel strategy––targeting signaling pathways that control ABC transporters at the BBB––to protect the brain, improve brain drug delivery, and reduce CNS pathology.
Introduction
The blood-brain barrier (BBB) is the interface between blood and brain that resides within the capillary endothelium and controls what goes into and comes out of the central nervous system (CNS). This barrier is highly active, dynamic, selective, and it responds to signals from the periphery and brain in both health and disease. A critical component of active barrier function is a group of ATP-binding cassette (ABC) efflux transporters that protect the brain from xenobiotics, including a myriad of therapeutics, by denying them access to the CNS. In addition to this physiological “gatekeeper” role, emerging evidence suggests that ABC transporters are also implicated in CNS pathology. For example, increased expression of BBB ABC efflux transporters may, in part, cause antiepileptic drug resistance in refractory epilepsy and thus contribute to uncontrolled seizures. In brain cancer, ABC transporters confer resistance to chemotherapeutics at the level of the BBB and in tumor cells in the patient and possibly in tumor stem cells. Moreover, ABC transporters may contribute to tumorigenesis. The role of ABC transporters in neurodegenerative disorders such as Alzheimer’s disease (AD) is also intriguing because they may hold part of the key to understanding the pathogenesis of this disease.
These new research findings call for novel and innovative scientific approaches and therapeutic strategies. One such approach is targeting the intracellular signaling pathways and regulatory networks that control ABC transporters located at the BBB.
This review focuses on recently identified signaling pathways and summarizes transporter regulation in inflammation, during oxidative stress, and by nuclear receptors. This review also highlights examples of how ABC transporters can be targeted to improve the therapy of epilepsy, brain cancer, and AD.
The Blood-Brain Barrier (BBB)/Neurovascular Unit
The original concept of a BBB goes back to experiments by Paul Ehrlich in 1885 (1). In his studies, Ehrlich observed that intravenously injected water-soluble dyes stain all organs except the brain and cerebrospinal fluid. Other researchers confirmed this finding, and in 1900, Lewandowsky coined the term “Bluthirnschranke” [German for blood-brain barrier (2)]. As early as 1898, Biedl and Kraus posited that barrier function could reside within the brain capillaries (3), but this hypothesis was not widely accepted until the 1950s. Eventually in the late 1960s, Reese, Karnovsky, and Brightman unequivocally demonstrated the existence of a barrier based on the brain capillary endothelial tight junctions (4, 5).
Today, the so-called neurovascular unit is considered the basic element that underlies BBB structure and function (Figure 1) (6). As currently recognized, the neurovascular unit is a complex anatomical arrangement of at least four cell types––capillary endothelial cells, pericytes, astrocytes, and neurons––that are integrated into one complex that controls barrier function. Initially considered a passive endothelial barrier to solute diffusion, it is now clear that the BBB is a highly active and dynamic endothelial interface that executes many important functions (7). For example, the BBB plays a crucial role in mediating and translating peripheral signals directed to the brain and vice versa. The BBB also has an immunological function, separating the innate immune system of the CNS from the peripheral immune system by preventing immune cells and pathogens, such as most viruses and bacteria, from entering the brain.
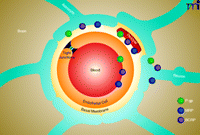
The blood-brain barrier (BBB)/neurovascular unit. The neurovascular unit comprises four cell types: capillary endothelial cells, pericytes, astrocytes, and neurons. The cells are integrated into one cell complex that controls critical BBB functions. The figure also shows the localization of the ABC drug efflux transporters P-glycoprotein (P-gp), BCRP (breast cancer resistance protein), and MRPs (multidrug resistance associated-proteins).
Lastly, the BBB executes a critical function by protecting the brain from xenobiotics. This barrier function of the brain capillary endothelium is based on three components: tight junctions, metabolizing enzymes, and selective transporters. Tight junctions represent the passive, physical component of the barrier and seal the paracellular spaces between adjacent endothelial cells, thereby restricting uncontrolled solute diffusion from blood to brain. Metabolizing enzymes, the biochemical component of barrier function, are considered a “second line of defense” because they inactivate xenobiotics that have entered the brain capillary endothelial cells. Lastly, transporters can be divided into two groups: 1) highly specific influx transporters including those of the solute carrier superfamily facilitate brain uptake of glucose, amino acids, ions, and other nutrients to meet the energy demand of the brain and 2) ATP-binding cassette (ABC) efflux transporters that represent the molecular basis of selective, active BBB function. ABC efflux transporters utilize ATP to actively clear the brain from metabolic wastes and to prevent xenobiotics, including harmful toxicants and a vast number of therapeutic drugs, from entering the brain. Therefore, ABC efflux transporters in the brain capillary endothelium are considered a “first line of defense” that protects the brain from xenobiotics.
ABC Transporters at the BBB
In 1989, two independent research groups detected the ABC efflux transporter P-glycoprotein (ABCB1, formerly known as MDR1) at the human BBB (8, 9). Several years later, P-glycoprotein was found at the BBB in mouse, rat, cat, dog, pig, cow, monkey, dogfish, and killifish (10–16). Since then, P-glycoprotein has been a main focus in the areas of BBB transporter and brain drug-delivery research. The importance of P-glycoprotein for barrier function and brain protection is best highlighted by experiments using genetic knockout mice. In vivo dosing studies using P-glycoprotein knockout mice show 5–50-fold increased brain-to-plasma ratios of a large number of therapeutic drugs that are P-glycoprotein substrates and normally cannot cross the BBB to enter the brain (17). Today, P-glycoprotein is considered the most prominent element of selective, active barrier function that limits xenobiotics from entering the brain.
Another ABC transporter at the BBB is BCRP (breast cancer resistance protein, ABCG2). Shortly after BCRP discovery in human and rat brain capillaries, it became clear that this transporter also plays a crucial role in brain-to-blood efflux of xenobiotics (18, 19). In this regard, BCRP and P-glycoprotein have partially overlapping substrate spectra. They cooperate to limit xenobiotics from entering the brain (20), and they compensate for one another (21). This cooperation and compensation are critical features that must be overcome for delivering chemotherapeutics across the BBB to treat brain tumors.
In addition to P-glycoprotein and BCRP, the multidrug resistance protein isoforms 1, 2, 3, 4, and 5 (MRPs 1–5; ABCC1–5) have also been identified at the BBB (22). MRPs are ABC efflux transporters that mainly transport organic anions, glutathione, glucuronide-or sulfate-conjugated compounds, and various nucleoside analogs. MRP1, 3, and 5 are highly expressed in brain tumors such as glioblastoma, in the tumor-supplying vasculature, and in the parenchymal tissue surrounding the tumor, where they confer resistance to chemotherapeutics (23); MRP1 has also been found in cancer stem cells (24). Furthermore, overexpression of MRPs in the brain capillary endothelium is associated with antiepileptic drug resistance in patients with epilepsy (25). Lastly, studies demonstrate that lack of MRP2 at the BBB results in increased brain drug levels, indicating a CNS protective role for this transporter (26).
The importance of P-glycoprotein, BCRP, and the MRPs for the BBB stems from four critical characteristics these transporters share: 1) substantial protein expression; 2) localization in the luminal plasma membrane of brain capillary endothelial cells, at the interface between blood and CNS; 3) highly effective and potent ATP-driven efflux transport against a concentration gradient; and 4) a remarkably broad substrate spectrum that covers a wide range of structurally diverse therapeutic drugs such as morphine, lapatinib, and cyclosporine A. These characteristics allow ABC drug efflux transporters at the BBB to protect the brain from toxicants while simultaneously restricting therapeutic drugs from entering the brain, and thus, impairing effective CNS pharmacotherapy. This observation poses a tremendous challenge for delivering drugs into the brain and limits successful treatment of CNS disorders.
Targeting ABC Efflux Transporters to Improve CNS Pharmacotherapy
Two strategies have been introduced to overcome efflux transporter-mediated, active, selective BBB function: transporter inhibition and modulation of transporter regulation.
Transporter Inhibition to Improve Brain Drug Delivery
By utilizing transporter inhibitors, one might overcome the function of the efflux transporter–mediated barrier and improve drug delivery to the brain. A large number of compounds have been screened for their ability to inhibit the major BBB ABC drug efflux transporters P-glycoprotein, BCRP, and MRPs; several potent inhibitors for each transporter have been identified (27). Results of animal studies testing these compounds were encouraging, and phase I and phase II clinical studies showed promise for some inhibitors (28, 29). Phase III trials, however, were disappointing and had to be terminated because of severe side effects and deaths (30). These trials demonstrate that in humans transporter inhibitors seem to have low potency, weak effectiveness, and poor selectivity, and would have to be given chronically at high doses to block transporter function effectively. However, such a therapeutic regimen bears an increased risk of severe side effects. Owing to these complications, no transporter inhibitors are currently in clinical use to improve brain delivery of CNS drugs.
Targeting ABC Transporter Regulation
Given the drawbacks of direct transporter inhibition, recent research has focused on elucidating the intracellular signaling pathways that control ABC efflux transporters at the BBB. The rationale for this approach is that finding the molecular switches of these transporters will allow selective modulation of transporter function and/or expression for therapeutic purposes in different clinical scenarios. Such an approach has three advantages. First, although direct transporter inhibition leaves little control over the extent and duration of barrier opening, targeting transporter regulation allows more subtle changes in transporter activity so transporters can be turned off for brief, controlled periods of time. Exploring this approach provides a time window during which the barrier is open to deliver normally nonpenetrating CNS drugs and reduces the risk of harmful toxicants entering the brain. Second, direct inhibition that blocks transporter activity can only be used to improve brain drug delivery. Targeting transporter regulation, on the other hand, can also be used specifically to increase transporter expression and/or activity to increase barrier function for therapeutic purposes. Such a strategy provides the opportunity to enhance brain protection and minimize central side effects during treatment of peripheral diseases. For example, the so-called “chemo brain” is a chemotherapy-induced cognitive dysfunction characterized by cognitive impairment that occurs in 20–30% of patients undergoing chemotherapy to treat cancer in the periphery. Therefore, treatment of patients with compounds that increase expression levels and activity of BBB efflux transporters prior to chemotherapy has the potential to reduce central side effects caused by anticancer drugs. Third, BBB ABC efflux transporters are affected by and contribute to CNS disease pathology (31, 32). Studies show that the efflux transporters P-glycoprotein, BCRP, and MRPs are involved in CNS disorders such as epilepsy, brain cancer, and AD where their role extends beyond that of solely extruding drugs. In such cases, direct transporter inhibition will not be of therapeutic benefit, whereas targeting the signaling pathways that control these transporters could be a useful therapeutic strategy. Thus, understanding the signaling pathways through which BBB ABC efflux transporters are regulated provides opportunities to protect the brain during treatment of peripheral diseases, to improve brain drug delivery to treat CNS disorders, and to prevent pathogenesis or slow the progression of CNS diseases.
Recently, several signaling pathways and regulatory networks that control efflux transporters have been identified at the BBB. We will first discuss basic signaling pathways that involve inflammatory mediators, oxidative stress, and nuclear receptors that have not been linked to one specific brain disorder. We will then focus on three specific pathways that regulate brain capillary efflux transporters in epilepsy, brain cancer, and AD.
Regulation of ABC Transporters by Inflammatory Mediators
Brain inflammation is involved in a variety of CNS disorders such as multiple sclerosis, stroke, brain cancer, epilepsy, and AD and Parkinson’s diseases (31). The release of pro-inflammatory cytokines in these CNS diseases triggers profound changes in gene expression in the brain and the BBB including changes in expression of ABC drug efflux transporters. The inflammatory mediators that have been studied most for their effect on efflux transporters are tumor necrosis factor-α (TNF-α), interleukin-1β(IL-1β), interleukin-6 (IL-6), interferon-γ (IFN-γ), and endothelin-1 (ET-1), and best described are their effects on P-glycoprotein. One of the first in vivo studies, by Goralski et al., shows that intracranial microinjection of lipopolysaccharide into rats to trigger cytokine-induced inflammation decreases P-glycoprotein expression in the brain, resulting in increased brain levels of the P-glycoprotein substrate digoxin (33). Consistent with this, intraperitoneally administered lipopolysaccharide inhibits BBB P-glycoprotein function in mice (34).
In vitro studies addressing the mechanism of BBB transporter regulation in inflammation demonstrate that TNF-α, IL-1β, IL-2, IL-6, and IFN-γ decrease P-glycoprotein expression and/or transport function in human brain endothelial cell lines (35, 36). Other groups, however, report an increase in transporter expression and function (37, 38). Hence, the first studies addressing BBB ABC transporter regulation in inflammation are inconclusive. This alleged contradiction is not surprising because inflammation is a complex process. These studies suggest instead that cytokine-mediated changes in P-glycoprotein expression and/or transport activity are time-, dose-, and location-dependent. For example, exposing isolated brain capillaries to nanomolar concentrations of TNF-α for a short period of time (one hour) rapidly decreases P-glycoprotein transport activity within minutes (39, 40). In this pathway (Figure 2A), TNF-α signals through the TNF receptor 1 (TNFR1), which triggers the release of ET-1 followed by signaling through the endothelin receptor B (ETB) and downstream activation of nitric oxide synthase (NOS) and protein kinase C (PKC). In contrast, long-term (six hour) exposure of brain capillaries to TNF-α increases P-glycoprotein expression and transport activity (Figure 2B). TNF-α signals through the same pathway (TNF-R1, ET-1, ETA/ETB, NOS, PKC), which, at longer exposures, involves both ETA and ETB as well as the transcription factor nuclear factor–kappa B (NF-κB) (41–43). Thus, P-glycoprotein transport activity is reduced after short exposure to TNF-α but increased with longer exposure times, indicating a complex, time-dependent regulatory mechanism. These findings suggest that brief activation of this signaling pathway could possibly be used to rapidly decrease P-glycoprotein transport activity, providing a window-in-time for delivering therapeutic drugs into the brain.

Regulation of BBB ABC transporters by inflammatory mediators and oxidative stress. (A and B) Time-dependent regulation of P-glycoprotein through a signaling pathway that involves the pro-inflammatory mediator tumor necrosis factor-α (TNF-α), TNF-α receptor 1 (TNF-R1), endothelin-converting enzyme (ECE), endothelin-1 (ET-1), endothelin A and B receptors (ETA/B), nitric oxide synthase (NOS), protein kinase C (PKC), and nuclear factor-κB (NF-κB). (C) Regulation of P-glycoprotein through a signaling pathway that involves the reactive oxygen species hydrogen peroxide (H2O2), extracellular signal-regulated kinase 1/2 (ERK1/2), stress-activated protein kinase (SAPK), protein kinase B (Akt), and protein kinase C (PKC). (D) Regulation of P-glycoprotein through a signaling pathway that is triggered by DEP (diesel exhaust particles) and involves NADPH oxidase (PHOX), reactive oxygen species (ROS), TNF-α-converting enzyme (TACE), TNF-α, TNF-R1, c-Jun N-terminal kinase (JNK), and activator protein-1 (AP-1).
In a recent proof-of-principle study, Rigor et al. demonstrate that in vivo activation of this TNF-α signaling pathway reduces P-glycoprotein transport activity, which enhances brain uptake of verapamil (44). Another study focuses on the effect of vascular endothelial growth factor (VEGF), which can be induced by TNF-α (45). This study shows that VEGF mediates rapid and reversible loss of BBB P-glycoprotein transport activity in isolated brain capillaries and that signaling involves the VEGF receptor flk-1 and the protein tyrosine kinase Src (46). Thus far, little is known about the regulation of other ABC drug efflux transporters at the BBB during inflammation. A study by von Wedel-Parlow et al. shows that TNF-α and IL-1β both decrease BCRP protein expression and function in brain capillary endothelial cells (47), which was previously demonstrated with ET-1 treatment (41). Poller et al. also demonstrate suppression of BCRP by IL-1β, IL-6, and TNF-α in a human BBB cell culture model (48).
These studies show that brain inflammation leads to profound changes of drug efflux transporters at the BBB and that complex, context-dependent regulation underlies the direction (increase or decrease) and extent of these changes. Given that brain inflammation is a major component of most CNS disorders, future research will likely identify more pathways where pro-inflammatory mediators signal changes in BBB transporter expression and/or function. Such knowledge could potentially be used to improve drug delivery into the brain.
Regulation of ABC Transporters by Oxidative Stress
Brain inflammation is often accompanied by oxidative stress, which participates in ischemia and neurodegenerative disorders such as AD and Parkinson’s disease. Recent studies show that in response to cellular stress both microglia and brain capillary endothelial cells can be major sources of reactive oxygen species, which affect ABC drug efflux transporters, and thus, BBB function (31, 49).
The first evidence for transporter regulation by oxidative stress at the BBB comes from in vitro experiments with hydrogen peroxide. Exposure of primary rat brain endothelial cells to hydrogen peroxide (for 24–48 hours) increases expression and transport activity of P-glycoprotein but not of Mrp1 (50). Nwaozuzu et al. confirm these findings and demonstrate that hydrogen peroxide–activated signaling through extracellular signal-regulated kinase 1/2 (ERK1/2), stress-activated protein kinase (SAPK), protein kinase B (Akt), and protein kinase C (PKC) increases P-glycoprotein expression and function (51) (Figure 2C).
Oxidative stress is also implicated in ischemia and stroke. Increased expression of P-glycoprotein, BCRP, and Mrp5 is observed in the periinfarct region following reversible middle cerebral artery occlusion in rats (52), and Spudich et al. show increased P-glycoprotein expression (3–24 hours) after focal cerebral ischemia in a mouse model (53). Moreover, this study demonstrates that pharmacological inhibition or genetic knockout of P-glycoprotein enhances the accumulation and efficacy of P-glycoprotein substrates such as FK506 and rifampicin in the ischemic brain.
In addition to brain inflammation, environmental toxins––such as diesel exhaust particles (DEP), a solid, small particulate matter air pollutant––also cause oxidative stress. DEP enter the body through the lungs and reach the brain (54, 55), where they trigger production of pro-inflammatory cytokines and reactive oxygen species (ROS). When isolated rat brain capillaries are exposed to DEP, P-glycoprotein expression and functional activity increases (49). This effect is mediated by NADPH oxidase activation and production of ROS, which initiates TNF-α converting enzyme (TACE)-mediated TNF-α release and signaling through TNF-R1, c-Jun N-terminal kinase (JNK), and activator protein-1 (AP-1) (Figure 2D). DEP exposure also increases expression of BCRP and several Mrp transporters, but it is not clear whether the identified signaling pathway is responsible for increased expression of all these ABC transporters or if other pathways may also be involved.
Lastly, other studies show that glutathione (GSH) depletion, which is involved in the pathogenesis of neurological disorders, increases oxidative stress in the brain that affects endothelial cells (56). Chronic oxidative stress induced by GSH depletion during CNS disease increases P-glycoprotein expression and transport function at the BBB in vitro and in vivo (57, 58).
These findings imply that oxidative-stress-mediated increases of drug efflux transporter expression tighten the BBB, which reduces drug penetration into the brain, and as a result, reduces efficacy of CNS drugs.
Regulation of ABC Transporters by Nuclear Receptors
In recent years, several nuclear receptors have been detected at the BBB including the pregnane X receptor (PXR), the farnesoid X receptor (FXR), and the constitutive androstane receptor (CAR). These nuclear receptors control the body’s defense, elimination, and excretory systems, that is, metabolizing enzymes and efflux transporters, in the liver, kidney, and intestine (59). It is now understood that these ligand-activated nuclear receptors also regulate enzymes and transporters in the brain capillary endothelium. Nuclear receptors are transcription factors that are activated by ligand binding, which initiates transcription of their target genes. PXR, FXR, and CAR are activated by endogenous compounds such as steroids, bile acids, and xenobiotics, including a large number of therapeutic drugs. At the BBB, nuclear receptor activation increases ABC transporter expression, which tightens barrier function (Figure 3). This phenomenon is demonstrated in three recent studies. In the first study, transgenic mice expressing human PXR are treated with the PXR activator rifampicin, which increases P-glycoprotein expression and transport activity in brain capillaries and results in reduced central antinociception of the P-glycoprotein substrate methadone (60). This finding suggests that increased P-glycoprotein reduces methadone uptake in the brain and, consequently, reduces the central methadone effect. In the second study, the PXR activator pregnenolone-16α-carbonitrile and the FXR ligand chenodeoxycholic acid increase BBB expression of the ABC transporter Mrp2 and the phase II metabolizing enzyme GSTπ (glutathione-sulfotransferase π) (61). This finding suggests coordinated nuclear receptor regulation of metabolism and transport at the BBB, comparable to what has previously been demonstrated in the liver. In the third study, Wang et al. report that exposure of isolated rat brain capillaries to the CAR activators phenobarbital and TCPOBOP {1,4-bis[2-(3,5-dichloro-pyridyloxy)] benzene} increases transport activity and protein expression of P-glycoprotein, BCRP, and Mrp2 (62). In vivo experiments with TCPOBOP confirm these findings and show increased protein expression and transport activity of all three transporters. Consistent with this observation, increased transporter expression is not observed in isolated capillaries from dosed CAR knockout mice (62).
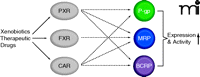
Regulation of BBB ABC transporters by nuclear receptors. Xenobiotic- and therapeutic-drug–mediated activation of the nuclear receptors pregnane X receptor (PXR), farnesoid X receptor (FXR), and constitutive androstane receptor (CAR) increases the expression of multiple ABC transporters at the BBB, creating a regulatory network.
These three studies show that nuclear receptor activation at the BBB increases brain capillary–localized ABC transporter expression and activity, resulting in barrier tightening. Because polypharmacy is a given in the clinic, PXR-mediated drug-drug interactions are a realistic scenario and could possibly be the basis for severe complications. This phenomenon is best highlighted by reports from transplant patients taking St. John’s Wort, an over-the-counter herbal antidepressant, in combination with the immunosuppressant cyclosporine A (63–65). The main constituent of St. John’s Wort is hyperforin, a potent human PXR activator that increases expression of metabolizing enzymes and drug efflux transporters, leading to altered pharmacokinetics of coadministered drugs (66). Accordingly, St. John’s Wort reduces cyclosporine A below therapeutic levels in patients, which results in transplant rejections. Indeed, of the multiple medications used by patients on a daily basis, some could activate nuclear receptors at the BBB, leading to increased ABC transporter expression, barrier tightening, and reduced brain uptake of CNS drugs. Certainly, drug-drug interaction at the level of the brain capillary endothelium would pose a major problem for CNS pharmacotherapy. On the other hand, nuclear receptor–mediated BBB tightening could be exploited for therapeutic purposes to protect the brain by selectively increasing expression of efflux transporters. This approach could be useful to prevent CNS side effects during treatment of diseases in the periphery (mentioned above). Selective up-regulation of BBB transporter expression via nuclear receptor activation could also be a useful strategy for brain disorders characterized by diminished transporter expression and/or activity. One such example is AD, in which BBB P-glycoprotein expression is reduced (discussed further below).
It is currently unknown whether changes in nuclear receptor–mediated transporter expression play a role at the BBB in humans, whether such changes could cause drug-drug side effects in the brain capillaries in humans, or whether this approach could be used therapeutically. Accumulating clinical data indicates, however, that nuclear receptors participate in the regulation of ABC transporters in intestine, liver, and kidney. It is possible that this will also be the case at the human BBB.
Targeting ABC Transporters in Epilepsy
Epilepsy is the most common serious neurological brain disorder and affects more than sixty million people worldwide. The majority of epileptic patients can be treated with antiepileptic drugs (AEDs), but despite the advances in drug development and the introduction of novel and effective AEDs in the past fifteen years, about 40% of patients––more than twenty million people, including two million children––respond poorly to AED pharmacotherapy (67, 68). These patients suffer from uncontrolled seizures that can vary in frequency (from few seizures per month up to thirty seizures per day) and duration lasting between seconds to hours (69). Because uncontrolled seizures can cause brain damage, patients experiencing prolonged seizures (status epilepticus) need immediate and effective treatment. However, therapeutic failure owing to AED resistance puts these patients in a life-threatening situation (70, 71). Consequently, patients with drug-resistant epilepsy have a sevenfold higher mortality compared to the general population or epileptic patients who respond to pharmacotherapy (72). In general, nonresponsive patients experience a low quality of life; despite advances in pharmacotherapy and neurosurgery, drug-resistant epilepsy remains a major clinical problem (73).
The cause for AED resistance in epilepsy is not yet fully understood, but various hypotheses exist. The multidrug transporter hypothesis of epilepsy posits that the number of ABC drug transporters, such as P-glycoprotein, BCRP, and MRPs, is increased at the BBB and might impart AED resistance (61, 74, 75). This hypothesis is based on the following findings. First, P-glycoprotein, BCRP, and MRPs are overexpressed in brain capillaries and in epileptogenic brain tissue from AED-resistant patients (76). Second, transporter inhibition increases brain uptake of several AEDs in animal seizure models (77). And third, prevention of seizure-induced transporter expression improves AED efficacy in drug-resistant epilepsy animal models (78). Moreover, recent studies imply that P-glycoprotein, BCRP, and MRPs might work in concert to prevent AEDs from entering the brain. Furthermore, ABC transporter overexpression at the BBB has been connected to increased seizure occurrence (79). This body of evidence has led to thorough investigation of the mechanism responsible for ABC transporter up-regulation at the BBB in epilepsy and a signaling pathway has been mapped for the induction of P-glycoprotein expression. In this pathway, seizures induce neuronal and glial release of glutamate, which signals through the N-methyl d-aspartate receptor (NMDA receptor), cyclooxygenase-2 (COX-2), prostanoid E receptor 1 (EP1), and NF-κB, resulting in increased expression of P-glycoprotein in brain capillaries (80–84) (Figure 4A). Consistent with this signaling pathway, seizure-induced P-glycoprotein up-regulation can be blocked by treating rats with celecoxib, a specific COX-2 inhibitor (84), and with SC-51089, a specific EP1 inhibitor (85). Importantly, van Vliet et al. demonstrate that COX-2 inhibition prevents induced P-glycoprotein expression and enhances brain uptake of phenytoin in rats with recurrent seizures. Schlichtiger et al. show that pretreatment with celecoxib restores the anticonvulsant efficacy of phenobarbital in AED-resistant rats that do not exhibit a significant response to phenobarbital prior to celecoxib treatment (78).
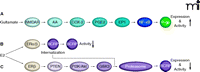
Targeting BBB ABC transporters in epilepsy and brain cancer. (A) Seizure-induced glutamate release up-regulates P-glycoprotein via a signaling pathway that involves the NMDA receptor (NMDAR), arachidonic acid (AA), cyclooxygenase-2 (COX-2), prostaglandin E2 (PGE2), the prostanoid E1 receptor (EP1), and the transcription factor NF-κB (nuclear factor-κB). E2 (17β-estradiol) signals reduction of BCRP expression (B) and its functional activity through a nongenomic signaling pathway that involves ERα/β, (C) the phosphatase and tensin homolog (PTEN, a tumor suppressor), phosphoinositide 3-kinase (PI3K), protein kinase B (Akt), glycogen synthase kinase 3 (GSK3), transporter internalization, and proteasomal degradation.
These studies provide a “proof-of-principle” that targeting signaling proteins involved in seizure-induced P-glycoprotein up-regulation may be a promising strategy to control transporter expression, increase brain levels of AEDs in the epileptic brain, and enhance AED efficacy to reduce seizure occurrence and frequency. It remains to be determined if these findings can be translated into the clinic. COX-2 inhibitors have been linked with severe side effects; thus, they will likely not be the first drugs of choice. Other proteins in the signaling pathway could potentially serve as better therapeutic targets to selectively modulate regulation of ABC transporters in epilepsy.
Targeting ABC Transporters in Brain Cancer
ABC transporters are best known for their contribution to multidrug resistance in cancer, where they prevent anticancer drugs from entering tumor cells through active efflux. Such transporter-mediated drug resistance limits the therapeutic benefit of chemotherapy. Research initially focused on inhibiting single transporters, but recent in vivo studies using transporter knock-out mouse models show that BCRP and P-glycoprotein work in concert at the BBB and possibly also in brain cancer cells and brain cancer stem cells to limit penetration of chemotherapeutics (20, 86, 87). These are tremendous clinical problems because successful treatment of brain tumors depends on chemotherapeutics to reach effective concentrations in the brain to eradicate metastases and brain tumor stem cell remnants that cannot be removed by surgery or irradiation.
Beyond their role in drug resistance, ABC transporters may also be involved in tumorigenesis (88). Tumors have an inflammatory microenvironment that stimulates tumor cell proliferation and that promotes angiogenesis and metastasis, thereby ensuring cell survival. ABC transporters might help create this inflammatory microenvironment by transporting pro-tumorigenic factors such as prostaglandins, leukotrienes, cyclic nucleotides, and platelet-activating factor out of cancer cells. Once in the extracellular fluid, these molecules are thought to bind to their extracellular G protein–coupled receptors via autocrine or paracrine signaling and sustain cancer-related inflammation (88). This mechanism is not yet fully understood, and the exact role ABC transporters play in tumorigenesis needs to be elucidated. However, based on observations that ABC transporters are highly expressed in tumors and tumor stem cells, that they potently confer chemotherapy resistance, and that they may be involved in tumor generation, proliferation, and survival, these transporters may be major targets in cancer therapy. Thus, identifying pathways that can be specifically targeted to decrease the expression of ABC transporters in cancer will be an important step toward improving chemotherapy of cancer, especially brain tumors.
In one such pathway, BCRP transport function in isolated brain capillaries is rapidly reduced by nanomolar concentrations of 17β-estradiol (E2) (89). Detailed dose-response experiments of 0.18 nmol/L, indicating that E2 is more result in an E2 EC50 potent in reducing capillary BCRP transport function than well-known BCRP inhibitors such as GF120918, fumitremorgin C, or Ko143. E2-mediated reduction of BCRP transport activity occurs within minutes, is reversible, and does not involve transcription, translation, or proteasomal degradation, indicating a non-genomic mechanism. Experiments using pharmacological inhibitors for estrogen receptor (ER) α and β, as well as experiments with ERα and ERβ knockout mice, demonstrate that E2 signals through both receptors to decrease BCRP transport function (Figure 4B). Extended, six-hour exposure of brain capillaries ex vivo to E2 or dosing mice with E2 leads to reduced BCRP expression and functional activity (90). Additional experiments reveal that E2 signaling involves ERβ phosphatase and tensin homolog (PTEN, a tumor suppressor), phosphoinositide 3-kinase (PI3K), Akt, and glycogen synthase kinase 3 (GSK3), and eventually results in internalization and proteasomal degradation of BBB BCRP (Figure 4C). Signaling through PTEN–PI3K–Akt regulates BCRP transport activity in glioma tumor stem-like cells (91). These data indicate that blocking the E2-signaling pathway reduces BCRP transport activity at the BBB and in cancer stem cells, suggesting that such an approach could potentially be used to increase brain uptake of chemotherapeutics that are BCRP substrates and improve their anticancer efficacy. Future studies will determine whether targeting ABC transporter regulation is also a viable strategy to interfere with their ability to transport tumorigenic factors and inhibit tumor angiogenesis, metastasis, and survival.
Targeting ABC Transporters in Alzheimer’s Disease (AD)
AD is a devastating brain disorder and AD patients decline mentally and physically and transform from functioning human beings into care-requiring dependents (92). There is currently no cure, no marker for early diagnosis, and no therapeutic intervention to prevent the disease, to alleviate disease symptoms, or to slow disease progression. AD is a complex neurodegenerative disease characterized by accumulation of neurotoxic amyloid β (Aβ) in the brain. Aβ is a small peptide that is generated by cleavage of amyloid precursor protein (APP); the most common Aβ isoforms in AD are Aβ40 and Aβ42. In healthy individuals, Aβ brain levels are low, but in AD patients, Aβ can be increased up to 100-fold in the brain, where it aggregates and forms plaques (93). These Aβ plaques are a hallmark for AD pathology and contribute to neuronal degeneration, memory loss, and dementia (93).
Recent reports indicate that Aβ brain accumulation, in part, arises from a failure to clear Aβ from the brain across the capillary endothelium into the blood (94, 95). This Aβ clearance mechanism must be a two-step process where Aβ must first pass through the abluminal (brain-side) and then the luminal (blood-side) plasma membranes of the brain capillary endothelial cells (Figure 5). Because Aβ is a 39–43 amino acid peptide, both steps must be facilitated by receptors and/or transporters. At the abluminal membrane, low-density lipoprotein receptor-related protein 1 (LRP1) seems to be the major protein responsible for the first step of Aβ uptake from the brain into capillary endothelial cells (94). Increasing evidence indicates that the primary protein at the luminal membrane mediating the critical second step––Aβ efflux from endothelial cells into blood––is P-glycoprotein. First, cell lines transfected with human P-glycoprotein can transport Aβ (96, 97). Second, Aβ brain clearance is reduced in P-glycoprotein null mice, and P-glycoprotein inhibition increases Aβ brain levels in a transgenic AD mouse model (98). Third, brain capillary P-glycoprotein levels are significantly reduced in postmortem brain samples from individuals with high Aβbrain deposition (99, 100). And fourth, BBB P-glycoprotein levels decrease with age, which could be one explanation why advanced age is associated with the highest risk for AD (11, 101).
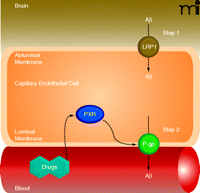
Targeting BBB P-glycoprotein in Alzheimer’s disease (AD). Proposed two-step Aβ clearance mechanism involving LRP1 (low-density lipoprotein receptor-related protein 1) in the abluminal membrane and P-glycoprotein in the luminal membrane of brain capillary endothelial cells. Ligand-activation of PXR restores blood-brain barrier P-glycoprotein in an AD mouse model and lowers Aβ brain levels.
Consistent with these observations, results from a recent study show that P-glycoprotein transports Aβ in isolated rat and mouse brain capillaries (102). This study also demonstrates that P-gp expression and transport activity are reduced by about 70% in brain capillaries from a transgenic AD mouse model [Aβ-overproducing human amyloid precursor protein (hAPP) mice], suggesting a link between high Aβ levels and reduced brain capillary P-glycoprotein in AD. Importantly, treating hAPP mice for seven days with pregnenolone-16α-carbonitrile, a specific activator for the nuclear receptor PXR (pregnane X receptor), fully restores BBB P-glycoprotein expression levels and decreases Aβ brain burden by up to 65%. Furthermore, results of a recent clinical trial show that rifampicin, a human PXR activator, improved cognition in AD patients over the twelve-month period of the study (103).
In addition to P-glycoprotein, BCRP also affects the accumulation of Aβ in the brain (104, 105). Xiong et al. demonstrate that BCRP transports Aβ in BCRP-overexpressing cell lines, that Aβ accumulates in the brain of BCRP knockout mice, and that BCRP expression is increased in brain samples from AD patients. The latter result, however, is contrary to what one would expect if BCRP is involved in Aβ clearance from the brain. These studies were conducted with postmortem brain samples from demented AD patients and with late-stage, cognitively impaired AD mice. In contrast, the study by Hartz et al., where no evidence was found for BCRP-mediated Aβ transport in brain capillaries, was conducted in twelve-week-old hAPP mice, at an age prior to cognitive impairment (102). This suggests that BCRP may not be involved in early stages of AD but may play a role in advanced stages of the disease. At this point, however, the role of BCRP in AD remains largely unclear and needs further investigation.
Together, these findings indicate that BBB transporters might be critical in AD onset and/or progression, which offers exciting opportunities for new treatments. In particular, restoring reduced BBB P-glycoprotein to normal expression levels in AD could be a novel therapeutic strategy to enhance Aβ brain clearance, thereby lowering Aβ brain load, delaying onset, and slowing progression of AD. However, because PXR regulates many target genes, PXR activation might not be the best approach to restore BBB P-glycoprotein therapeutically. Targeting other signaling pathways might therefore provide better alternatives to restore brain capillary P-glycoprotein in AD.
Conclusions
ABC efflux transporters at the BBB restrict delivery of drugs into the brain, which severely impairs pharmacotherapy of CNS disorders. Beyond this role in drug efflux, it is now clear that ABC transporters are also affected by and contribute to CNS pathology. This new paradigm provides opportunities for novel therapeutic strategies and requires innovative approaches to modulate BBB transporters for treatment purposes in different clinical scenarios. Unraveling intracellular signaling pathways and networks, and identifying molecular switches that regulate ABC transporters at the BBB, will provide new molecular targets for CNS therapy. Thus, targeting ABC transporter regulation at the BBB will help protect the brain, improve brain drug delivery, and prevent and slow progression of CNS pathology.
Acknowledgments
We thank Emma Soldner and Emily Madole for editorial assistance. This work was supported by the Whiteside Institute for Clinical Research and the Duluth Medical Research Institute (to AH and BB), and by an AACP New Investigator Award and a UMN GIA Award (both to BB).
- Copyright © 2010
References

Anika M.S. Hartz, PhD, is Assistant Professor in the College of Pharmacy, University of Minnesota. She holds a BS in pharmacy from the University of Heidelberg, Germany, and received her doctorate in pharmaceutical sciences/pharmacology from the same institution; her doctoral work concerned with the regulation of P-glycoprotein at the blood-brain barrier. Dr. Hartz continued her work as a postdoctoral fellow at the NIEHS/NIH and then as a Research Associate in the Department of Biochemistry and Molecular Biology, Medical School, University of Minnesota. She works on efflux transporter regulation in Alzheimer’s disease and brain cancer.

Björn Bauer, PhD, is Assistant Professor in the College of Pharmacy at the University of Minnesota. He received a BS in pharmacy and a PhD in pharmaceutical sciences/pharmacology from the University of Heidelberg, Germany, and then conducted work as a Postdoctoral Fellow at the NIEHS/NIH in Research Triangle Park, NC. Dr. Bauer’s research focuses on the intracellular regulation of blood-brain barrier ABC transporters in CNS disease. E-mail bjbauer{at}d.umn.edu; fax 218-726-6500.

