Emerging Roles for PKC Isoforms in Immune Cell Function
The protein kinase C (PKC) family consists of thirteen members categorized as conventional, novel, atypical, or PKC-related isoforms, depending on whether diacylglycerol (DAG), calcium, or phosphatidylserine (PS) is required for their activation (1,2). Mice deficient in the conventional PKC isoform PKCβ are immunodeficient, exhibiting a loss of peritoneal B-1a B cells and reduced T-cell–independent antibody responses (3). B cells from PKCβ-deficient mice also fail to respond to BCR stimulation, suggesting that PKCβ is a critical component of the BCR signaling machinery. Similarly, mature T lymphocytes in PKCθ-deficient mice exhibit defects in T cell antigen receptor- (TCR)-induced proliferation and reduced T-dependent responses (4). These results demonstrate the critical role for specific PKCs in B and T cell receptor signaling and function.
In contrast to the immunodeficiencies observed in PKCβ and PKCθ mice, two recent reports demonstrate that mice deficient in the novel PKC isoform PKCδ exhibit B cell hyperactivation, which leads to autoimmunity (5,6). Whereas early B cell development in the bone marrow of PKCδ-deficient mice is normal, exaggerated B cell expansion occurs in the spleen and other peripheral organs, resulting in splenomegaly and lymphadenopathy. The presence of excessive serum autoantibodies specific for DNA and nuclear proteins led to immune-complex deposition and glomerulonephritis in PKCδ–/– mice. Furthermore, transplantation of PKCθ-deficient B cells into normal animals (i.e., adoptive transfer) indicated that this hyperproliferative phenotype was B cell specific (5), underscoring the essential role for PKCδ in negative regulation of B lymphocyte signaling.
The autoimmune phenotype of PKCδ–/– mice was associated with a loss of peripheral tolerance. For example, the B cells of mice that carry a BCR transgene that specifically recognizes hen egg lysozyme (IgHEL) will undergo negative selection (i.e., activation-induced cell death) in the periphery of transgenic mice that have been bred with transgenic mice expressing soluble hen egg lysozyme (sHEL) (7,8). However, negative selection observed in doubly transgenic IgHEL–sHEL mice was abrogated in the absence of PKCδ, suggesting a critical role for PKCδ in regulating peripheral B cell tolerance (6).
The mechanisms responsible for the breakdown of tolerance in PKCδ–/– B cells remain unclear. Peripheral B lymphocytes develop in a step-wise fashion from immature transitional 1 (T1) to transitional 2 (T2) to naïve, follicular mature B cells (9). T1 and T2 cells reside in distinct splenic microenvironments and exhibit differential responsiveness to BCR engagement whereby T1 B-cells might be the target for peripheral negative selection, and T2 B-cells might be the target for BCR-dependent positive selection (10). Evaluation of the signaling functions of PKCδ specifically within T1 vs. T2 peripheral B cell populations might therefore provide important insight into how PKCδ functions to regulate peripheral B cell tolerance.
Notably, the data from Miyamoto et al. suggest that PKCδ may regulate B cell activation by inhibiting interleukin (IL)-6 production. IL-6 expression and secretion are increased in PKCδ–/– splenic B cells (5). This is consistent with the exaggerated B cell expansion and plasmacytosis observed in IL-6 transgenic mice (11). IL-6 expression is dependent on the transcriptional activator NF-IL6, or the CCAAT/enhancer-binding protein (C/EBPβ). PKCs can phosphorylate NF-IL6 in vitro on inhibitory residue Ser240, leading to a marked decrease of NF-IL6 DNA-binding activity (12). Lipopolysaccahride- (LPS)-induced NF-IL6 DNA binding activity was greatly increased in PKCγ–/– B cells (5), suggesting that PKCδ may regulate B cell tolerance, in part, by inhibiting IL-6 production through site-specific phosphorylation of NF-IL6.
Interestingly, PKCβ appears to possess a similar ability to phosphorylate critical inhibitory residues on Bruton’s tyrosine kinase (Btk). PKCβ is activated upon BCR stimulation, in response to increased concentrations of DAG and Ca2+. Sustained generation of these two second messengers is mediated by Bruton’s tyrosine kinase (Btk) through the activation of PLC-γ isoforms (Figure 1⇓) (13). Although PKCβ has a definite role in mediating the positive effects downstream of Btk, PKCβ (and possibly other conventional PKCs) can also inhibit Btk through a negative feedback loop (14). PKCβ specifically phosphorylates Btk on Ser180 within the Tec homology (TH) region, leading to the inhibition of Btk membrane translocation and activation (14). In addition, the atypical PKC isoform PKCμ (or PKD) also negatively regulates BCR signaling through the phosphorylation-dependent inhibition of the tyrosine kinase Syk (15). Therefore, the ability to negatively regulate lymphocyte signaling by site-specific phosphorylation at inhibitory residues may be a common characteristic of many PKC family members (Figure 1⇓).
In addition to the negative effects mediated by PKCδ on lymphocyte activation, an earlier study using chemical inhibitors suggested that PKCδ functions in a positive role in BCR dependent NF–κB activation (16). In contrast to these findings, the reports by Miyamoto et al. and Mecklenbräuker et al. demonstrate that NF–κB activation in B cells is completely intact in PKCδ-deficient mice (5,6). These differences in results emphasize that caution should be maintained when using inhibitors with partially overlapping specificity to assess the physiologically relevant function of proteins.
In contrast to the intact NF–κB function in PKCδ–/– mice, recent studies demonstrate that PKCβ is the major PKC isoform required for BCR-dependent NF–κB activity. Splenic B cells from PKCβ-deficient mice exhibit severe defects in survival, Bcl-xL induction, IκB degradation, and IκB kinase (IKK) activity in response to BCR engagement (17). The mechanism of NF–κB regulation by PKCβ appears to be mediated through PKCβ-dependent recruitment of IKK into the membrane lipid rafts that are associated with the BCR signaling complex. In contrast, CD40-dependent NF–κB activity is fully intact in PKCβ–/– B cells, suggesting that this role for PKCβ is specific to BCR signaling. Similar to the role for PKCβ in B cells, TCR-dependent activation of NF–κB is abrogated in splenic T cells from PKCθ–/– mice (4), suggesting an analogous function for PKCθ in regulating TCR-dependent recruitment of IKK into lipid rafts and NF–κB activation. Together, these findings suggest that PKCβ and PKCθ are responsible for cell-specific and receptor-specific activation of the NF–κB signaling pathway in B and T lymphocytes, respectively (Figure 2⇓).
Implicit in the identification of specific roles for individual PKC isoforms in positive or negative regulation of lymphocyte signaling is that PKCs may represent useful potential drug targets for various immune disorders. For example, the autoimmune disease observed in PKCδ–/– mice suggests that negative regulatory functions by PKCs are crucial for maintaining proper immunological tolerance. It remains to be seen whether PKCδ function is reduced in certain autoimmune states, and whether specific activators of PKCδ can be developed to treat such conditions. Furthermore, because PKCδ and PKCβ seem to have negative and positive roles, respectively, in B lymphocytes, it will be interesting to test whether concomitant PKCβ-deficiency might abrogate the autoimmunity observed in PKCδ–/– mice.
Chemical activators such as TPA (12-O-tetradecanoylphorbol-13-acetate) are not specific for individual PKCs, nor are PKC isoform-specific activators available (18,19). In contrast, a number of inhibitors specific for individual PKCs do exist including antisense oligonucleotides and chemical inhibitors of catalytic activity (18). The positive roles for PKCβ and PKCθ in lymphocyte function suggest that highly specific inhibitors of these PKC isoforms may have great potential in treating B-cell– or T-cell–specific disorders. Although no PKCθ-specific chemical inhibitors exist, several PKCβ-specific inhibitors are currently available, including the macrocyclic bis (indolyl) maleimides LY-333531, LY-379196 and LY-317615 (20). These compounds are well tolerated systemically and can be administered orally (21). The availability, safety, and efficacy of PKCβ-specific oral inhibitors suggest that they may also be useful in the treatment of certain B-cell immune disorders. For example, crossing Btk-deficient mice with lupus-prone (NZB x NZW)F1 mice abrogates the systemic lupus erythematosus (SLE)-like symptoms in these animals (22). Similar experiments involving the breeding of PKCβ–/– mice to (NZB/W)F1, to PKCδ–/–, or to other autoimmune-prone mice will be required to validate PKCβ as a drug target for B-cell–dependent autoimmune diseases. In addition, a clinically refractory subset of non-Hodgkin’s diffuse large B cell lymphomas (DLBCLs) exhibit elevated PKCβ expression (23). Recent studies in our laboratory demonstrate efficacy for PKCβ-inhibitors in specifically blocking the survival of PKCβ-expressing DLBCL tumor lines in vitro (17).
Further studies will be needed to confirm the efficacy of PKCβ-specific inhibitors in treating B cell malignancies and autoimmune disease in vivo. The future development of PKCθ-specific inhibitors and PKCδ-specific activators may similarly lead to effective treatments for T cell lymphomas and various autoimmune conditions. Therefore, understanding the positive and negative regulation of PKC function in the immune system may lead to effective treatments for human disease in the near future.
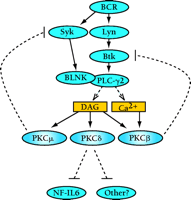
Negative roles for PKC isoforms in B lymphocyte signaling. B cell receptor (BCR) engagement leads to a cascade of signaling events, eventually leading to the activation of the B cell. Specific PKC isoforms that are activated by the BCR can inhibit B cell signaling. Although PKCβ and PKCμ function in negative feedback loops to inhibit the upstream activators Btk (itself activated bt the tyrosine kinase Lyn) and Syk, respectively, PKCδ may function to inhibit the NF-IL6 transcription factor. BLNK, B cell linker protein; Syk, a 72-kDa protein tyrosine kinase highly expressed in B cells; Btk, Bruton’s tyrosine kinase; DAG, diacylglycerol; NF-IL6, nuclear factor for interleukin-6 expression.
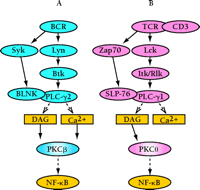
Positive roles for PKC isoforms in lymphocyte signaling. Engagement of the BCR (A) or the T cell antigen receptor (TCR) (B) activates a set of similarly acting signaling molecules. Specific Src (Lyn and Lck), Syk-Zap70, and Tec (Btk and Itk/Rlk) family kinases are activated. These events mediate increased activity of PLC-γ, leading to the generation of the second messengers DAG and calcium, which are required for the activation of most PKCs. Both PKCβ and PKCθ, respectively, function in controlling BCR and TCR dependent NF–κB activation. SLP-76, src homology (SH)2 domain–containing leukocyte protein of 76-kD.
- © American Society for Pharmacology and Experimental Theraputics 2002
References

Beichu Guo, MS, is a PhD student in the Department of Immunology at the University of Washington, and is a member of Rawlings laboratory.

Thomas T. Su is an MD-PhD student in the School of Medicine and the Molecular Biology Institute, University of California, Los Angeles, and is a member of Rawlings laboratory.

David J. Rawlings, MD, is the Head of the Section of Pediatric Immunology/Rheumatology at the University of Washington and Children’s Hospital Medical Center, Seattle; and Associate Professor of Pediatrics and Immunology, in the School of Medicine at the University of Washington. Address comments to DJW. E-mail: drawling{at}u.washington.edu

