Modulating Modulation: Crosstalk Between Regulatory Pathways of Presynaptic Calcium Channels
The central nervous system expresses a range of different voltage-gated calcium channels (termed N-, P/Q-, L-, R- and T-types), each with unique subcellular distributions and specialized functions. The N- and P/Q-types are largely found in presynaptic nerve terminals where they are both physically and functionally involved in neurotransmitter release. The modulation of N- and P/Q-type channels by a variety of G protein-coupled receptors and associated intracellular messenger cascades is consequently a key mechanism for regulating synaptic transmission. The activation of presynaptic calcium channels is inhibited by direct interactions with G protein βγ (Gβγ ) subunits (1, 2) . This phenomenon was first described more than two decades ago (3) , but it was not until more recently that we have begun to understand the underlying molecular details, and to appreciate its complex nature ( for review, see 4) . We now know that Gβγ subunits bind to the cytoplasmic-region linking domains I and II of the N- and P/Q-type calcium channel α1 subunits (5, 6) (Figure 1⇓), that N- and P/Q-type channels are differentially regulated by distinct types of Gβ subunits (7–9) , and that calcium channel subunit composition regulates G protein efficacy (10, 11) .
In contrast to the action of Gβγ subunits, the activation of protein kinase C (PKC) through either the metabotropic glutamate receptor-subtype 1 (mGluR1) or by application of phorbol esters mediates an increase in N-type current activity in in vitro heterologous expression systems and in neurons (6, 12, 13) . A number of additional protein kinases including cGMP-dependent protein kinase (14) and mitogen-activated protein (MAP) kinases (15) also modulate N-type and P/Q-type channel activity. One unique aspect of the action of PKC is that phosphorylation of a single residue, Thr422 , in the linker region between domains I and II of the N-type calcium channel, antagonizes subsequent G protein–dependent inhibition of the channel (6, 12, 16, 17) . The crosstalk that occurs between the G protein and PKC pathways is dependent on the nature of the Gβ subunit isoform (18) and may account for the observation that the channel inhibition mediated by different types of G protein-coupled receptors varies in PKC sensitivity (12, 18) .
In addition to second messenger control, presynaptic calcium channel activity is regulated by interaction with synaptic vesicle release proteins. For example, the binding of syntaxin-1A to the domain II-III linker of the N- or P/Q-type calcium channels reduces the availability of the channel for opening (19–23) . Interestingly, this regulatory mechanism intersects with second messenger regulation in two ways: First, PKC-dependent phosphorylation of the syntaxin binding site on the N-type calcium channel antagonizes the action of syntaxin (23, 24) . Secondly, and perhaps more intriguingly, syntaxin-1A regulates G protein-mediated inhibition of N-type calcium channels (21, 23, 25) . The fact that calcium influx through P/Q-type calcium channels triggers syntaxin-1A expression (20) raises the possibility that P/Q-type channels could also indirectly control the activity of N-type channels. Taken together, there appears to be an extensive interaction between individual types of regulatory pathways (Figure 1⇓), most of which occur directly at the level of the calcium channel α1 subunit.
The level of complexity of calcium channel modulation has increased yet further with a recent article by Wu et al. (26) that demonstrates a novel type of regulation of presynaptic calcium channels by phosphatidylinositol-4,5-bisphosphate (PIP2 ). Direct modulation of ion channels by PIP2 was first reported for ATP-dependent K+ (KATP ) channels (27) and was subsequently demonstrated for other types of potassium channels (28, 29) and transient receptor potential (TRP) channels (30) . The report by Wu et al. is, however, the first to reveal that PIP2 has an effect on any type of calcium channel. The authors show that the time-dependent decrease in current activity (rundown) of P/Q-type calcium channels following membrane patch excision could be slowed and partially reversed by the addition of PIP2 , and accelerated with antibodies against PIP2 . Interestingly, PIP2 also strongly inhibited the activation of P/Q-type channels, resulting in a ∼30 mV depolarizing shift in the voltage dependence of activation. This latter effect is reminiscent of the action of Gβ subunits, but it remains unclear as to whether the effects of Gβ subunits and PIP2 are additive or work through a common mechanism. Of particular note, the authors show that activation of adenosine 3’,5’-monophosphate-(cAMP)-dependent protein kinase (PKA) antagonized the inhibitory effect of PIP2 . Based on these findings, the authors proposed the presence of regulatory and stabilization domains capable of interacting with PIP2 , with the regulatory domain being a target for PKA-dependent phosphorylation. This model is similar to that proposed for PKC- and G protein-mediated crosstalk discussed above. However, in the absence of site-directed mutagenesis data on the channel complex itself, at this point it remains unclear whether the effects of either PIP2 or those of PKA occur directly at the level of the channel or through an intermediary. Wu et al. also examined native N-type calcium channels in bullfrog sympathetic neurons. Using luteinizing hormone releasing hormone (LHRH) to effect the hydrolysis of PIP2 , the authors showed that PIP2 appears essential for stabilizing N-type channel activity. This effect mirrors the protective effect of PIP2 against P/Q-type channel rundown, but unlike with P/Q-type channels, the authors did not report a second, inhibitory action of PIP2 on N-type channel function. Nonetheless, the data clearly show that both types of presynaptic calcium channels are regulated by PIP2 , suggesting yet another layer of complexity in the regulation of presynaptic calcium homeostasis.
It thus appears that there are at least two sets of regulatory systems that fine tune presynaptic calcium channel activity. On one hand, there is an intricate interplay between G proteins, PKC, and syntaxin, and on the other hand, there is crosstalk between PKA and PIP2 modulation of channel activity. It seems likely that there may be additional interplay between the two regulatory systems. PIP2 is the precursor for IP3 and diacylglycerol, both of which are involved in the activation of PKC. Regulating PIP2 concentrations could therefore conceivably affect the activity of some isoforms of PKC, and potentially, its crosstalk with G protein- and syntaxin-dependent regulation of the channels. Why do nerve cells need such a complex web of regulatory mechanisms converging on the calcium channel molecule? The notion that the individual regulatory pathways intersect at the level of the channel protein rather than simply regulating each other’s activities per se ensures that crosstalk is specific for the target channel. Thus, the integration of multiple regulation pathways with different time courses ranging from milliseconds, for direct Gβγ -dependent inhibition, to minutes, for kinase activity, allows for the precise temporal control of, and maximum specificity for, calcium influx and consequently neurotransmitter release.
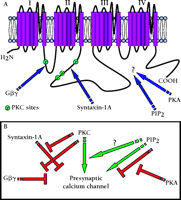
Proposed transmembrane topology of the calcium channel α1 subunit, illustrating target sites for the PKC-dependent phosphorylation, syntaxin binding, and G protein-dependent modulation. A. The sites of action of PIP2and PKA remain to be identified, and it remains possible that their effects are mediated by interactions with one of the ancillary calcium channel subunits.B. Crosstalk between regulatory pathways converging on presynaptic calcium channels. Inhibitory (red) and stimulatory (green) interactions are shown.
- © American Society for Pharmacology and Experimental Theraputics 2002
References

Terrance P. Snutch, PhD, is a professor in the Biotechnology Laboratory at the University of British Columbia and a Senior Scientist of the Canadian Institutes of Health Research (CIHR). Email snutch{at}zoology.ubc.catel. (604) 822-6968 fax (604) 822-6470

Gerald W. Zamponi, PhD, is an Associate Professor in the Physiology and Biophysics Department at the University of Calgary, a CIHR Investigator and AHFMR Senior Scholar, and the holder of the Novartis Chair for Schizophrenia research. Email zamponi{at}ucalgary.ca

