Mechanism of Tissue-Selective Drug Action in the Cardiovascular System
- 1Department of Physiological Systems, Johnson & Johnson Pharmaceutical Research & Development L.L.C., San Diego, CA;
- 2School of Pharmacy and Pharmaceutical Sciences, SUNY at Buffalo, Buffalo, NY;
- 3Departmentent of Pharmacology & Therapeutics, University of British Columbia, Vancouver BC Canada;
- 4Department of Pharmacology & Toxicology, Queen’s University, Kingston ON Canada
Abstract
Analysis of the human genome project tells us that there may be as few as 3000 genes that are likely to be good drug targets. Although the number of targets is still very large, these data have been interpreted by some to mean that the pharmaceutical industry may someday run out of novel drug targets. Despite the doom and gloom of such analysis, there is considerable reason for optimism. Drugs may exhibit selectivity of action beyond that predicted by target expression alone. Drugs that act at a single molecular target may have very different pharmacology and, as a result, different therapeutic uses. Three well-characterized model systems are highlighted to illustrate this point. The first model system is exemplified by nifedipine and verapamil, both of which act on L-type calcium channels. Both drugs are used to treat hypertension, but only verapamil can be used to produce atrioventriclar block in patients with atrial fibrillation. The second model system describes the therapeutic exploitation of unusual conditions that occur in the ischemic myocardium to produce drugs that are more effective for suppressing ischemia-induced arrhythmias. The third model system discusses the mechanisms through which phosphodiesterase-5 (PDE5) inhibitors act selectively to facilitate penile erection while having little effect in the non-penile vasculature that also expresses PDE5.
Introduction
Pharmacologists often envision a simple model of drug action in which the agonist drug binds to a receptor expressed on the cell surface, activates it, and this results in a measurable change in the cell’s properties. This model describes the drug’s specific action (1); however, drug action must also be considered in the context of the whole organism, and selectivity of action can be considered the second defining feature of a drug.
The mechanism through which drugs achieve selectivity of action can be as simple as the presence of the receptor on the responsive tissue and not on other tissues. In this model, the drug’s effect is governed by its affinity for the receptor, its ability to activate that receptor (i.e., its efficacy), and the abundance of the receptor on the target cell. A classic experiment described by Furchgott (2) illustrates the point nicely. Incubation of the irreversible antagonist dibenamine produces a parallel, rightward shift in the dose-response curve for histamine in guinea pig ileum. Subsequent incubations with dibenamine continue to shift the concentration-response curve to the right, and with further exposure, the maximum response is reduced. Physiology simply occurs in the reverse direction. As receptor number increases, the tissue becomes sensitive to the agonist. The magnitude of the response is increased to some maximum; the tissue then becomes more sensitive to the drug such that the same maximum response is achieved at lower concentrations of the agonist. Reversible antagonists behave as described for dibenamine except that the maximum response to the agonist drug is not reduced (except under special conditions). These systems behave as though the receptor density is reduced by the antagonist drug.
Having drugs that do one thing but not another is at the heart of pharmacology and therapeutics (1), and a discussion of drug action is not complete without considering its selectivity. A drug’s selectivity can then be defined as the ratio of the drug concentration required to produce each of the drug’s specific actions. Although receptor selectivity is certainly a great way to achieve selective drug action, it is equally clear that drugs can exhibit selectivity of action that cannot be explained simply on the basis of preferring one molecular target over another.
The concept that drug distribution influences drug selectivity predates the receptor concept [reviewed by Limbird (3)]. Paul Ehrlich described accumulation of lead in the brain and used this observation to explain its preferential action on the central nervous system. It follows that accumulation of the drug at the site of action is one possible mechanism through which drugs can exhibit selectivity of action. Conversely, accumulation of the drug at the site of toxicity can be expected to reduce the selectivity and thereby the drug’s therapeutic utility. Modern drug delivery technologies exploit this mechanism to make products that act more selectively and are, as a result, safer.
Three examples where drugs have been demonstrated to have tissue selective actions in the cardiovascular system are highlighted to illustrate mechanisms whereby drugs exhibit selectivity of action beyond the paradigm of receptor density.
Cardiovascular-Specific Calcium Channel Blockers of Diverging Structure
The calcium channel blockers (represented in Figure 1⇓ by the first-generation drugs diltiazem, nifedipine, and verapamil) are a structurally, pharmacologically, and therapeutically heterogeneous group of drugs that have achieved prominence both as therapeutic agents and as chemical tools with which to probe channel structure, function, and localization [for general reviews see (4, 5)]. These agents, and their second- and third-generation 1,4-dihydropyridine analogs, exhibit significant differences in pharmacological profile and therapeutic application, despite interacting at a common target, namely, the L-type voltage-gated calcium channel (Table 1⇓). This channel comprises one class of a larger family of voltage-gated calcium channels that includes the high voltage–activated L-, N-, P/Q-, and R-type channels and the low voltage–activated T-type channel (5, 6). Each of these calcium channels is a heteromeric association of subunits, of which the α1 subunit forms the ion pore, contains the drug-binding sites, and thus determines the major properties of the channel. Each channel class has distinct biophysical and pharmacological properties, shows distinctive localization, and can be further divided into subclasses (e.g., splice variants).
Calcium Channel Antagonists: Effects on Physiological Parameters and Therapeutic Use
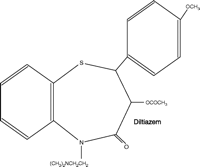
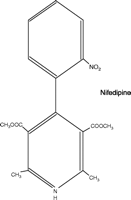
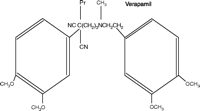
Structures of diltiazem, nifedipine, and verapamil.
Tissue Selectivity
Differences in tissue selectivity can exist for drugs not only across the three major classes of calcium channel blockers, but also within a given class; Table 2⇓ indicates the wide variation in selectivity evident among the 1,4-dihydropyridines. These differences are therapeutically important and can arise in principle from any of a number of factors, several of which are discussed below [for general reviews, see (4, 5)].
Cardiovascular Selectivitya of Calcium Channel Antagonists
Mode of Calcium Mobilization
Most excitable and non-excitable cells maintain a variety of calcium mobilization processes to support stimulus–response coupling. The calcium channel blockers under consideration, however, exert their actions (at therapeutic concentrations) primarily, if not exclusively, at L-type channels. Because respiratory smooth muscle contraction is not mediated by calcium influx through L-type channels, these blockers are ineffective in asthmatic states. Vascular smooth muscle can be remarkably heterogeneous in its sensitivity to these blockers; whereas both afferent and efferent renal arterioles contract in response to angiotensin, only the former are sensitive to the blockers.
Class and Subclass of Channel
L-type currents are mediated by multimeric complexes that contain the α1S, α1C, α1D, or α1F subunit isoforms; these, resepectively, are dominantly expressed in skeletal muscle (CaV1.1), cardiac and smooth muscle (CaV1.2), neuroendocrine (CaV1.3), and retinal cells. Currents mediated through the α1C subtype in cardiac and smooth muscle exhibit the highest sensitivity to the therapeutically employed calcium channel blockers. Splice variants of the CaV1.2 channel exist, including the cardiac 1.2a and the vascular smooth muscle 1.2b forms, and although detailed comparative pharmacology is not available, the smooth muscle form is more sensitive to the 1,4-dihdyropyridine nisoldipine (7, 8).
Location of the Drug-binding Site
Early studies, largely employing radioligand binding techniques, established that diltiazem, the 1,4-dihydropyridines, and verapamil occupy distinct, or partially distinct, binding sites allosterically coupled to each other. Labeling and molecular biology studies have identified specific residues on three regions of the α1 subunit that are involved in binding. Diltiazem and 1,4-dihydropyridine bind at the extracellular entry to the channel, whereas the verapamil site is located at the intracellular surface (6).
State-dependent Interactions between Drug and Channel
The affinity of calcium channel blockers for the channel is determined by the state of the channel—resting, open, or inactivated—over the course of the excitation cycle (9). Whereas verapamil and diltiazem block currents in a frequency-dependent manner (i.e., increased frequency of depolarization promotes blocking), nifedipine and other 1,4-dihydropyridines exhibit voltage-dependent interactions (i.e., maintained depolarization promotes blocking); verapamil is thus regarded as preferentially binding to (or accessing preferentially) the open state of the channel, and the 1,4-dihdyropyridines thus appear to favor the inactivated state. A good correlation has been shown between vascular selectivity and binding to the depolarized states by a series of clinically available dihydropyridines (10). These observations underlie the antiarrhythmic properties of verapamil and the general vascular selectivity of the 1,4-dihdyropyridines.
Pharmacokinetics and Efficacy
Pharmacokinetic factors can profoundly alter drug selectivity. Rapid bolus administration of nifedipine produces a larger increase in cardiac rate, compared to slow administration, because of reflex activation due to the relative speed in the fall of blood pressure (11). Pharmacokinetic factors can determine differential selectivity between vascular beds. Thus, nimodipine exhibits cerebral blood vessel selectivity, in part because it has a much higher transfer rate, relative to nifedipine, across the blood–brain barrier (12).
Channelopathies
There is an increasing awareness of functional disorders associated with mutations in calcium channels—termed “channelopathies.” Mutations in the L-type channel are associated with several disorders of human skeletal muscle, including muscular dysgenesis, hypokalemic periodic paralysis, and malignant hyperthermia. Congenital stationary night blindness is associated with mutations in the CaV1.4 channel, and a variety and mutations in the CaV1.2 channel are associated with multi-system defects, including autism and arrhythmias (13).
Exploitation of Pathological Conditions to Achieve Selective Antiarrhythmic Actions
The ideal of creating drugs that are exclusively therapeutic (and not toxic) is rarely met. The techniques of molecular biology, high-throughput screening, and combinatorial chemistry are all used to try and achieve this goal with varying degrees of success. Certain approaches to making drugs that are tissue-specific seek to take advantage of characteristics that are particular to the target tissue (e.g., antibiotics). A further approach involves prodrugs that are selectively released, or activated, at their site of action.
Selectivity may also be achieved by exploiting conditions that are particular to the disease state. For certain arrhythmias, there are specific pathological conditions that might be exploited for therapeutic advantage. Many arrhythmias are, in effect, tachycardias, and so drugs that are selectively active at high heart rates (i.e., positive frequency dependence) have obvious advantages. If specific ion channels are activated by arrhythmogenic pathological conditions, and this activation results in arrhythmias, then blockade of such channels could be expected to be selectively antiarrhythmic. New antiarrhythmic drugs might alternatively function to target the production or action of arrhythmogenic mediators; however, such approaches have not been overwhelmingly rewarding. Some arrhythmias are produced by characteristic pathological changes in cardiac tissue. Thus, ventricular fibrillation is most common in the setting of acute ischemia, and the ischemic tissue itself is intimately involved in arrhythmogenesis. Conditions in ischemic tissue are very different from those in normal tissue and might thus be selectively targeted. Indeed, ischemia involves both a fall in pH and an increase in extracellular potassium ion concentrations, conditions that might be utilized to confer selectivity of action upon antiarrhythmic drugs.
Selective Antifibrillatory Actions in the Setting of Myocardial Ischemia
Clinical Observations of Antifibrillatory Potential
For many years, lidocaine was considered a safe and effective drug for the prevention of ventricular fibrillation in the setting of acute myocardial infarction. It was hypothesized that lidocaine was therapeutically more valuable than other sodium channel–blocking antiarrhythmics because of its state-dependent binding to the channel conformation (inactivated state) that is more common in partially depolarized myocytes and tissue stimulated to beat at high frequencies. Thus, lidocaine and other Class Ib antiarrhythmics should have a natural selectivity for ischemic arrhythmias and, provided that sodium channel blockade confers antifibrillatory actions, be clinically useful. However, this does not appear to be so, and the current dominant view is that, although lidocaine may produce a limited reduction in the incidence of ventricular fibrillation in the clinic, it is liable to evoke asystole.
Beta-blockers reduce mortality in patients who have had a myocardial infarct. Such clinical benefit is, however, limited, and we are currently unsure as to whether this protective action is due to antiarrhythmic or other mechanisms. The only other drug that possibly prevents ventricular fibrillation clinically is amiodarone. It has been shown, in some instances, to produce a limited reduction in mortality in patients considered at risk of ventricular tachycardia or fibrillation, whether or not in the setting of acute myocardial infarction. The mechanisms underlying the limited antifibrillatory actions of amiodarone are not fully understood as a consequence of its wide pharmacological spectrum of actions. Otherwise, interest in antifibrillatory drugs is currently low in academia and industry (14), primarily because conventional sodium channel–blocking drugs that were designed to prevent ventricular tachyarrhythmias increase, rather than decrease, mortality (15). Furthermore, research has still not clearly revealed the arrhythmogenic substrate responsible for ventricular fibrillation. This fact obviously hampers drug development. Drugs that target the Na+/H+ exchanger, KATP channels, or specific potassium channels in an attempt to achieve antifibrillatory activity have proved to be of very limited value [see (16)].
Novel Antifibrillatory Drugs: Future Directions
Because ventricular fibrillation is the greatest single cause of death in richer countries, the impetus to pursue the development of antifibrillatory agents should be great. Box 1 suggests that certain properties are required of an antiarrhythmic in order for it to act selectively against ventricular fibrillation without incurring excessive cardiovascular toxicity. Not everyone would agree with all the statements made in Box 1, but complete concurrence with them is not necessary in the following argument.
Looking at the Problem: Potential Features of Ventricular Fibrillation.
-
The major (but not sole) cause of ventricular fibrillation is probably acute sustained myocardial ischemia leading to infarction.
-
Acute sustained myocardial ischemic tissue is characterized by elevated extracellular potassium and hydrogen ion concentrations, as well as by other less well-defined changes.
-
The electrophysiological consequences of acute sustained myocardial ischemia include slowed conduction and altered refractoriness. These factors in and of themselves initiate and sustain ventricular fibrillation, presumably via re-entry mechanisms.
-
Ventricular fibrillation is initiated and sustained by disordered electrogenesis; electrical quiescence imposed upon ischemic tissue should prevent ventricular fibrillation.
-
Fibrillation, in effect a variant of tachycardia, is characterized by high-frequency action potentials.
Selective ion channel blockade must be implemented to the point of effectively inducing electrical quiescence in the ischemic zone if one is to prevent ventricular fibrillation occurring as a result of sustained myocardial ischemia. Assuming that ion channels in the ischemic zone have the same fundamental nature as those in normal tissue, we can concentrate on discovering drugs with selectivity of action in ischemia and with the appropriate positive frequency dependence. In addition, we can attempt to exploit the special conditions found in ischemic myocardium so as to potentiate ion channel blocking actions, or to activate an appropriate prodrug. Possible mechanisms for such activation might include acid dependent-hydrolysis, perhaps by recruitment of an endogenous acid-dependent enzyme. Such considerations form the basis of the postulates outlined in Box 2, which we have attempted to test in various ways.
Characteristics Sought in a Selective Antifibrillatory Drug.
What: A drug that blocks those channels (e.g., sodium, potassium channels and perhaps L-type calcium channels) responsible for initiating and maintaining ventricular fibrillation; the channel-blocking profile of amiodarone serves as an example.
Where: The agent should be selective for ischemic ventricular myocytes because ion channel blockade in normal tissue causes toxicity.
How: Blocking actions dependent on the elevated potassium and hydrogen ion concentrations found in ischemic ventricular tissue; as ischemia becomes more severe or protracted, so would the blocking actions.
On the basis of a large number of studies, we decided to concentrate our efforts on a series of substituted aminocyclohexylamide analogs, with the intention of developing compounds whose ion channel blocking actions were potentiated by the extracellular acid conditions and extracellular potassium conditions associated with ischemic tissue and were frequency dependent. An initial study by Bain et al. (17) compared two novel compounds with lidocaine to determine whether ischemia selectivity was provided by this approach. As suggested in Table 3⇓, both compounds (RSD1000 and RSD1019) effectively protect against ventricular fibrillation induced in an experimental in vivo model of acute sustained myocardial ischemia. The antiarrhythmic protection afforded by both compounds is better than that obtained with lidocaine, and they are no more likely than lidocaine to cause convulsions in conscious animals. When tested in vitro, RSD1000 and RSD 1019 block Kv4.2 and Kv1.5 channels as well as sodium channels. Blockade in sodium channels is frequency dependent.
Selective Drugs? Antiarrhythmic and Pharmacological Profiles
On the assumption that the channel-blocking activity of the RSD compounds depended on the degree of protonation of a tertiary nitrogen in the molecule, a series of compounds of varying pKa values was investigated for antiarrhythmic actions in ischemia and their ability to block cardiac ion channels under normal and acid conditions. The major results of that study (19), which included examination of RSD1000 and seven other related analogs, lidocaine, quinidine, and flecainide, show that the most effective antiarrhythmic against ischemia-induced arrhythmias were not necessarily the most potent, but they were the most potent in conditions of ischemia relative to potency in normal cardiac tissue. When the dose-response curves for antiarrhythmic activity against ischemia are compared for all the drugs, compounds vary markedly. Some compounds, such as RSD1000 and RSD1030, produce 100% abolition of arrhythmias and a very predictable dose-response curve, whereas others, which rank lowest in effectiveness (e.g., quinidine, flecainide, and RSD944) provide at most 20–30% of the maximum protection possible. Furthermore, the dose-response curves for the low-ranked compounds are aberrantly shaped (e.g., flat or bell-shaped). Not only does rank order of relative effectiveness for their antiarrhythmic doseresponse curves correlate positively with the rank order for relative ischemic selectivity, but it also negatively correlates with the propensity to produce unwanted side effects. These unwanted side effects include falls in blood pressure and heart rate but also depressive effects in normal cardiac tissue. Obviously, the therapeutic indices are also best for the compounds providing the best antiarrhythmic protection.
The success of implanted electrical defibrillators and related electronic devices has not obviated the need for drugs that will prevent the initiation and maintenance of ventricular fibrillation. As indicated above, there are a number of potential avenues that can be explored to provide drugs that will selectively abolish ventricular fibrillation due to acute sustained myocardial ischemia. By concentrating on the special condition of ischemia (e.g., elevated potassium and hydrogen ion concentrations) it has proved possible to produce experimental compounds that provide a degree of selective protection against ventricular fibrillation due to acute sustained myocardial ischemia, at least in an experimental model.
Drug Selectivity Determined by Tissue Function: Inhibition of PDE5 in the Penis
Erectile dysfunction (ED), defined as the consistent inability to maintain an erection sufficient for sexual intercourse, can have several etiologies. ED can be primarily caused by organic factors, such as vascular disease, diabetes, and surgery-related trauma, or by psychological factors. Comorbidities that contribute to ED severity include hypertension, cardiovascular disease, cerebrovascular accidents, peripheral vascular disease, hypercholesterolemia, and perineal or penile trauma (20, 21).
Erectile Physiology and Nitric Oxide
Briefly, the generation of an erection is a complex neurovascular event that involves central and peripheral nervous systems as well as the penile vasculature (Figure 2⇓). Visual, auditory, and tactile erectogenic stimuli are integrated and processed centrally in several hypothalamic structures. Once integrated, both branches of the autonomic nervous system recruit nerves innervating the erectile tissues of the penis (Figure 2⇓).

Mechanism of erection. [Modified with permission from International Education Initiative on Erectile Function Core Slide Kit, copyright Physicians World GmbH (2002).]
Structurally, the penis consists of three columns of erectile tissue: the corpus spongiosum, which contains the urethra, and two corpora cavernosa (Figure 3⇓). Each corpus cavernosum is composed of trabecular smooth muscle and connective tissue and sinuses that fill with blood during sexual stimulation, thereby facilitating penile erection. A thick tunica albuginea, composed of layers of collagen and elastin fibers, encases the corpora cavernosa and acts to compress corpora cavernosa veins during sexual stimulation, thereby limiting drainage and prolonging the erection (Figure 3⇓).
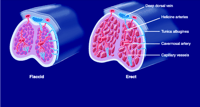
Structural composition of the penis. The penis consists of three columns of erectile tissue: the corpus spongiosum, which contains the urethra, and two corpora cavernosa, which contain sinuses and trabeculae. See text for details. [Modified with permission from International Education Initiative on Erectile Function Core Slide Kit, copyright Physicians World GmbH (2002).]
Nitric oxide (NO), the dominant vasorelaxant messenger of corpora cavernosa smooth muscle cells, is released from terminal branches of “nitrergic” corpora cavernosa nerves as well as from endothelial cells of the corpora cavernosa (Figure 4⇓) (20–22). Release of NO leads to relaxation of smooth muscle cells in the corpora cavernosa vasculature and allows rapid filling of the sinusoidal spaces and enlargement of the penis. In addition to increasing blood supply, smooth muscle cell relaxation also allows the sinusoids to become more compliant (20–22), thus assisting in the filling process. At a molecular level, relaxation of smooth muscle cells occurs through the NO-mediated activation of the soluble enzyme guanylyl cyclase (23), which synthesizes the second messenger cyclic guanosine 3′,5′-monophosphate (cGMP). Activation of a cGMP-dependent protein kinase (protein kinase G) leads to phosphorylation of several smooth muscle cell proteins that coordinate smooth muscle cell relaxation by causing smooth muscle cell hyperpolarization, closure of voltage-activated calcium channels, and sequestration of intracellular calcium (Figure 4⇓) (22–24).
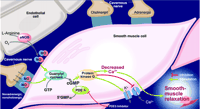
Physiological basis of corpora cavernosa smooth muscle relaxation. The NO–cGMP–protein kinase G cascade in the mechanism of penile erection is indicated. The role of PDE5 inhibition as a therapeutic target in ED is also indicated. [Modified with permission from International Education Initiative on Erectile Function Core Slide Kit, copyright Physicians World GmbH (2002).]
NO-mediated Treatment of ED
Alterations in corpora cavernosa arterial inflow, as well as venoocclusive disorders, can contribute to ED (1). At a molecular level, these processes are generally thought to be due to an inadequate relaxation of the corpora cavernosa smooth muscle cells caused by insufficient NO delivery to these tissues. Because the NO–cGMP–protein kinase G cascade is the major physiological pathway regulating smooth muscle cell relaxation in the corpora cavernosa, the supply of NO to the penis, along with the effectiveness of the NO that is released in corpora cavernosa tissues, is a potential therapeutic target in ED. Nevertheless, attempts to promote NO-mediated corpora cavernosa smooth muscle cell relaxation by way of local, or systemic, administration of NO-releasing vasoactive agents, such as nitrovasodilators, are undermined by the potential to cause systemic hypotension (21). Indeed, whereas hypertension can contribute to ED by limiting blood vessel dilatation, agents that decrease blood pressure contribute to ED by reducing perfusion pressure to the organ (20, 21, 23). In contrast, an approach based on potentiating the effects of endogenously released NO by inhibiting cGMP turnover in smooth muscle cells of the corpora cavernosa has been very successful. Indeed, several selective inhibitors of the major cGMP-hydrolyzing enzyme expressed in these cells, phosphodiesterase-5 (PDE5), are effective in treating ED in human males (22–24).
The effectiveness of PDE5 inhibitors in treating ED is based on sound predictions about the effect of limiting cGMP hydrolysis in tissues expressing significant levels of PDE5 (Figure 4⇑). Because PDE5 is expressed in most visceral and vascular smooth muscle cells (23), the molecular basis for the clinically observed “functional selectivity” of these agents in treating ED (i.e., without marked effects on systemic blood pressure) is perhaps less clear. Indeed, PDE5 inhibitors have been shown in vitro to alter cGMP-mediated cellular functions in numerous cell types.
Based on the work of several groups, it appears likely that two interdependent factors are at play in generating the clinically evident selectivity of PDE5 inhibitors in the penis. First, the neural circuitry integrating erectogenic stimuli and activating nitrergic nerves likely increases NO release selectively in the penis. In contrast, in non-penile tissue, in which the erectogenic stimuli do not increase nitrergic activity, NO release does not increase and cGMP levels thus remain low. Secondly, cGMP is not only the substrate of PDE5, but also an allosteric activator, so that PDE5 activity is elevated under conditions of increased intracellular cGMP concentration (25–28). In this way, nitrergic innervation and allosteric activation of PDE5 function specifically in the penis to create the conditions amenable to PDE5 “tissue-based inhibitor selectivity.” In non-penile tissues, in which there is no increased flux of cGMP, PDE5 inhibitors would be predicted to have no effect.
Conclusions
Cardiovascular pharmacology offers multiple examples of drug selectivity that goes beyond the parameters of receptor density and selectivity for molecular targets. Calcium channel antagonists can manifest selectivity by taking advantage of the electrophysiological characteristics of cardiac tissue; tissue selectivity can also be influenced by the chemical structure of the antagonist. In the context of myocardial ischemia, there remains a need for drugs that will prevent the initiation and maintenance of ventricular fibrillation. By concentrating on the special conditions of ischemia (e.g., raised extracellular potassium and acidic conditions), it has been possible to obtain experimental compounds that provide a degree of selective protection against ventricular fibrillation occurring as a result of these conditions. In males, a physiology-based mechanism of drug selectivity is exemplified through the functionality of PDE5 inhibitors for the treatment of erectile dysfunction; in this case, the unique physiology of the erectogenic response provides the specific conditions that engender drug selectivity.
Acknowledgments
This article stems from an Experimental Biology 2005 symposium sponsored by the Division of Systems and Integrative Pharmacology and the Division for Cardiovascular Pharmacology of ASPET. T.D. Barrett would like to thank Michael J. Curtis for the on-going discussion of specificity and selectivity and Guy Lagaud for fueling the idea for the symposium.
- © American Society for Pharmacology and Experimental Theraputics 2005
Selected References

Donald Maurice, PhD, is Associate Professor of Pharmacology & Toxicology at Queen’s University in Kingston, Ontario, Canada and Heart & Stroke Foundation of Ontario Career Investigator. Don has had a long-standing interest in PDE-mediated signaling in cell of the cardiovascular system. Currently, since the NHL season has been cancelled, Don is likely fishing.

Michael J.A. Walker, PhD, is Professor of Pharmacology & Therapeutics at the University of British Columbia, Vancouver, Canada. His interests include myocardial ischemia and the development of antiarrhythmic drugs.

David J. Triggle, PhD, is University Professor and a Distinguished Professor in the School of Pharmacy and Pharmaceutical Sciences at the State University of New York at Buffalo. His principal research interests include drug–receptor interactions and the chemical pharmacology of drugs active at ion channels.

Terrence D. Barrett, PhD, is Senior Scientist on the Physiological Systems Team at Johnson & Johnson Pharmaceutical Research and Development in San Diego. He organized the Experimental Biology 2005 symposium on which this article is based. Address correspondence to TDB at TBarret1{at}PRDUS.JNJ.COM.

