Discovery and Therapeutic Promise of Selective Androgen Receptor Modulators
Abstract
Androgens are essential for male development and the maintenance of male secondary characteristics, such as bone mass, muscle mass, body composition, and spermatogenesis. The main disadvantages of steroidal androgens are their undesirable physicochemical and pharmacokinetic properties. The recent discovery of nonsteroidal selective androgen receptor modulators (SARMs) provides a promising alternative for testosterone replacement therapies with advantages including oral bioavailability, flexibility of structural modification, androgen receptor specificity, tissue selectivity, and the lack of steroid-related side effects.

Introduction
The beneficial effects of testosterone on muscle, bone, and physique have been known for over a century. Yet testosterone and its esters are approved for only a limited number of therapeutic applications, including primary or hypogonadotropic hypogonadism and delayed puberty. Testosterone and structurally-related anabolic steroids have been demoted to the therapy of final resort for anemia, endometriosis, and metastatic breast cancer owing to the recent development and widespread clinical use of more effective therapies (e.g., erythropoietin, aromatase inhibitors, and taxanes). Recent interest in using testosterone as hormone replacement in aging men or in age-related frailty has been slowed because of widespread concerns related to the effects of testosterone on the prostate, serum lipids, and cardiovascular system. The discovery and clinical development of selective estrogen receptor modulators (SERMs) transformed the therapeutic use of estrogens. Nonsteroidal selective androgen receptor modulators (SARMs) with the ability to selectively stimulate or maintain muscle and bone mass with lesser pharmacologic effects in the prostate are now leading a similar revolution in the therapeutic use of androgens.
Action of Androgens on Target Tissues
The overall physiological effects of endogenous androgens are contributed by testosterone and its active metabolites, dihydrotestosterone (DHT) and estradiol. Approximately 6 to 8% of testosterone is converted to DHT through the action of type 2 5α-reductase, an enzyme highly expressed in male accessory sex organs, hair follicles, and genital skin. Approximately 0.3% of testosterone is converted to estradiol via the action of aromatase, an enzyme expressed in the brain, liver, and adipose tissue (1). Testosterone and DHT execute their actions predominantly through the androgen receptor (AR), which belongs to the nuclear receptor superfamily and functions as a ligand-dependent transcription factor. More than 95% of circulating testosterone is synthesized and secreted by the Leydig cells in the testes. Circulating testosterone is essential for the differentiation and growth of male accessory reproductive organs (e.g., prostate and seminal vesicles), control of male sexual behavior, and the development and maintenance of male secondary characteristics that involve muscle, bone, larynx, and hair. (1). For decades, androgens have been primarily used for hormonal replacement in hypogonadal men. Whereas severe hypogonadism is uncommon, disease and aging-related androgen insufficiency is much more frequent. Low endogenous testosterone concentrations are associated with sarcopenia and frailty arising from decreased fat-free mass, lessened muscle strength, and reduced bone mineral density (BMD). Recently, multiple clinical trials of hormone replacement using testosterone were conducted in aging men [for review, see (2)]. The potential benefits of testosterone replacement therapy include increase in BMD, improvement in body composition and strength, sexual function, cognitive function, and mood; however, the potential risks of such treatment, including those in the cardiovascular system, blood (e.g., hematocrit and hemoglobin levels), and prostate are routinely experienced. Large-scale and long-term clinical trials are needed to evaluate the risk–benefit ratio of testosterone replacement therapy in aging men. Another important line of research using testosterone is hormonally-mediated male contraception (referred to below, for brevity’s sake, as hormonal male contraception). A variety of attempts have been made to produce pharmacologic, effective, reversible and side effect–free contraceptive methods for the male. Hormonal male contraception has only recently reached the stage of clinical development [for review, see (3)].
Spermatogenesis
Circulating testosterone participates in the regulation of androgen production by the hypothalamus-pituitary-testis axis. As illustrated in Figure 1⇓, gonadotropin-releasing hormone (GnRH) is released from the hypothalamus and stimulates the pulsatile secretion of luteinizing hormone (LH) and follicle-stimulating hormone (FSH) from the pituitary. In the testis, LH indirectly stimulates spermatogenesis through testosterone synthesized by Leydig cells, while FSH directly interacts with FSH receptors expressed in Sertoli cells and stimulates spermatogenesis. Testosterone and its aromatized metabolite (estradiol) negatively regulate circulating levels of testosterone in the hypothalamus and pituitary. Activin and inhibin produced by Sertoli cells stimulate or inhibit, respectively, the secretion of FSH from the pituitary (4). High concentrations of intratesticular testosterone are essential for the initiation and maintenance of spermatogenesis as evidenced by the infertility of hypogonadal men. Results from both animal models and man, however, support the requirement for both FSH and testosterone in achieving quantitative and qualitative spermatogenesis [for review, see (5)].
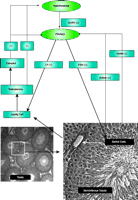
Hypothalamus-pituitary-testis axis of androgen regulation. The production of testosterone is regulated by hormones produced from hypothalamus and pituitary. (+) represents positive feedback and (-) represents negative feedback.
Bone
Bone is a living tissue, which is continuously being broken down (i.e., bone resorption) and regenerated (i.e., bone formation) by osteoclasts and osteoblasts, respectively. AR ligands affect BMD by changing overall osteoblastic activity and osteoclastic activity, resulting from changes in the total number of each cell type and individual cell functional capacity (6). These actions are mediated directly by the AR and by paracrine and autocrine action. The underlying mechanisms for paracrine and autocrine action have been reviewed in detail elsewhere (6). Androgens seem to have the ability to decelerate the bone remodeling cycle and tilt the focal balance of the cycle toward bone formation. The loss of androgens is thought to increase the rate of bone remodeling by removing restraining effects on osteoblastogenesis and osteoclastogenesis. Also, androgens exert dual effects on the lifespan of mature bone cells, with anti-apoptotic effects on osteoblasts and osteoclasts and pro-apoptotic effects on osteoclasts (7). DHT also stimulates osteoblast proliferation under experimental conditions (8). The effects on osteoblast differentiation are rather controversial, but most in vitro studies suggest that androgens have a stimulatory effect on expression of alkaline phosphatase, type I collagen, and osteocalcin, and increased mineralization of the extracellular bone matrix (9). Moreover, DHT has a suppressive effect on osteoclast differentiation (10).
Muscle
The mechanism of androgen action on muscle remains largely unknown. The common hypothesis is that androgens promote muscle protein synthesis. There is evidence to support the idea that testosterone supplementation increases muscle protein synthesis in elderly men (11) and young hypogonadal men (12). Also, androgen-induced increases in muscle mass appear to arise from muscle fiber hypertrophy rather than hyperplasia (i.e., cellular enlargement rather than cellular proliferation) (13). Androgen increases crosssectional areas of both type I and type II muscle fibers in a dose-dependent manner, but does not alter the absolute number or the ratio of type I and type II fibers. Androgeninduced increases in muscle fiber cross-sectional area were correlated with the increase in myonuclear number and satellite cell number. These findings suggest that androgen increases satellite cell number, resulting in muscle fiber hypertrophy and myonuclear number increase. The molecular mechanisms of the androgenic effect on satellite cell numbers are not well understood. Taylor et al. reported that androgen stimulates the differentiation of mesenchymal pluripotent cells to the myogenic lineage (14); however, other possible pathways, including increases in satellite cell proliferation and decreases in satellite cell apoptosis are possible but remain unknown.
Prostate
In the prostate, testosterone is rapidly converted to DHT by type 2 5α-reductase. Conversion to DHT amplifies the action of testosterone by 3–5 fold, owing to the greater binding affinity of DHT (as compared to testosterone) to the AR (15). DHT plays a critical role in determining prostate size prior to and during adulthood and is believed to be essential for the development of benign prostate hyperplasia (BPH), which occurs in 50% and 90% of men in their fifties and nineties, respectively, in the United States. The major problem associated with BPH is lower urinary tract symptoms (LUTS). Multiple lines of evidence suggest the importance of androgen, especially DHT, in the development of BPH. For instance, BPH does not develop in males with certain type 2 5α-reductase mutations or in males with very low levels of androgen due to prepubertal castration or hypopituitarism-related hypogonadism (16). Moreover, clinical treatment of BPH either by chemical or surgical castration, or with a type 2 5α-reductase inhibitor (e.g., finasteride) induces apoptosis of epithelial cells, which in turn significantly decreases the volume of the prostate (17). Recently, the role of age-dependent changes in the intraprostatic hormonal environment in the development of BPH was evaluated. Despite the aging-related decrease in testosterone and intraprostatic DHT production, an increased estradiol–DHT ratio was found in the transition zone of aging human prostate. This relative estrogen-dominant status was believed to be relevant to the development of BPH (18). Furthermore, estradiol is capable of inducing precancerous lesions and prostate cancer in aging dogs (19). Therefore, testosterone supplementation in older men raises concern with regard to acceleration of BPH and/or prostate cancer.
Limitations of Steroidal Androgens
Since the discovery of the therapeutic benefits of testosterone in the 1930s, a variety of androgen preparations have been introduced and used clinically. Unfortunately, virtually all of the current available androgen preparations have severe limitations (20). Unmodified testosterone demonstrates little pharmacologic activity after oral administration resulting from its rapid hepatic elimination. To prolong pharmacologic effects, testosterone implants and longer acting esters, including testosterone enanthate (TE), testosterone propionate (TP), testosterone buciclate (TB), testosterone undecanoate (TU), and testosterone decanoate (TD), were developed. Except for TU, the administration routes of most testosterone esters are limited to intramuscular injection (im), surgical implantation for implants and pellets, or transdermal delivery, such as patches and gels. Furthermore, serum testosterone levels fluctuate greatly between injections, and skin rashes and irritation are associated with testosterone patches. The other major limitations for using steroidal androgens for hormonal male contraception, which requires high doses of androgen, are steroid-related side effects, including decrease of HDL cholesterol, increase of hematologic parameters such as hemoglobin and hematocrit, increased body weight, and acne (3). To use testosterone for potential long-term hormone replacement in aging men, the potential risk in the prostate and cardiovascular system needs to be evaluated carefully by large prospective clinical trials.
Pharmacophores and Molecular Mechanism of SARMs
The recent and successful marketing and clinical application of selective estrogen receptor modulators (SERMs) stimulated a great interest in the discovery and development of nonsteroidal selective androgen receptor modulators (SARMs). Progress has been made in identifying novel pharmacophores of nonsteroidal SARMs by structural modification of nonsteroidal antiandrogens. As listed in Table 1⇓, SARM pharmacophores can be classified into four categories: aryl-propionamide, bicyclic hydantoin, quinoline, and tetrahydroquinoline analogs. One uniform characteristic of these compounds is that they are not substrates for aromatase or 5α-reductase. These nonsteroidal AR ligands are known to act as full agonists in anabolic organs (e.g., muscle and bone) but as partial agonists in androgenic tissues (e.g., prostate and seminal vesicles). Additionally, some SARMs have more favorable pharmacokinetic properties, AR receptor specificity, and are more amenable to structural modifications than their steroidal counterparts. SARMs may therefore be of benefit for the treatment of primary or secondary hypogonadism, osteopenia or osteoporosis, frailty, acquired immunodeficiency syndrome (AIDS) or cancer-related cachexia, rehabilitation, anemias, BPH, and hormonal male contraception.
Pharmacophores of SARMs
General molecular mechanisms of tissue-selective nuclear receptor modulators were proposed recently to facilitate understanding of nuclear receptor pharmacology (21). Within the nuclear receptor superfamily, molecular mechanisms of SERMs are best understood. The tissue selective action of SERMs is mainly based on the differential expression of two types of estrogen receptor (ER) in target tissues and consequently the differential ligand-ER conformation, the promoter context of the target genes, and recruitment and availability of coregulator proteins (22). In contrast to the ER, a single form of the AR is ubiquitously expressed throughout the body. Notably, testosterone is locally metabolized to DHT by type 2 5α-reductase, which is highly expressed in the prostate and other genital tissues but not in anabolic organs, such as muscle and bone. DHT is a more potent androgen than testosterone and is believed to amplify the androgenic activity of testosterone in some tissues. Administration of finasteride, a 5α-reductase inhibitor, to intact male rats significantly decreases the prostate mass. Additionally, administration of testosterone with a 5α-reductase inhibitor in castrated rats attenuated the androgenic activity of testosterone in the prostate (23). As with SARMs, which are not substrates for 5α-reductase, the tissue selectivities of testosterone and DHT arise from, at least partially, the lack of such active metabolic amplification in androgenic organs. Other possible molecular mechanisms related to the tissue selectivity of SARMs include ligand-dependent changes in AR conformation, differential interaction with the promoter context of target genes, and the differential recruitment of coregulators in target tissues. The discovery of SARMs not only provides a potentially significant therapeutic advance for androgen replacement therapy, but also provides model compounds to further study the molecular mechanism of action of the AR.
Outline of Drug Discovery of Aryl-Propionamide SARMs
Nonsteroidal AR agonists (i.e., androgens) were recently reported by our laboratories as well as others (24–27). Compounds that demonstrated higher anabolic activity than androgenic activity in vivo were identified as selective androgen receptor modulators (SARMs). The ultimate goal of research in this field is to discover chemical compounds that can be used for androgen replacement therapy to address one or some functions of prototypic steroidal androgens without unwanted side-effects. Treatment should be tailored to the specific need of patients with the best desirable pharmacologic activity. Although a variety of pharmacophores and lead molecules are being developed for clinical use (Table 1⇑), the majority of published preclinical research to date focuses on a series of aryl-propionamide analogs first reported in 1998 (24). Information regarding the stateof- art drug discovery of the aryl-propionamide SARMs is given in the sections below.
In Vitro and In Vivo Structure-Activity Relationships of Nonsteroidal AR Ligands
Early structure-activity relationship (SAR) work on hydroxyflutamide analogs (28) confirmed the importance of an electron-deficient aromatic A-ring and of the substituents attached to the carbon atom bearing a tertiary hydroxyl group. Two years later, Tucker et al. (29) reported that the AR binding and antiandrogenic activity of hydroxyflutamide and bicalutamide (Figure 2⇓) derivatives were optimum when the 4-position substituent in the A-ring was either a cyano or nitro group and the 3-position substituent was a chloro or trifluoromethyl group. It’s interesting to note that partial androgen agonist activity was observed in some trifluoromethyl-substituted compounds, suggesting that AR agonists could be designed and developed by subtle structural modification(s) of known AR antagonists. When the idea of trifluoromethylation was utilized by our laboratory to discover novel aryl-propionamide AR agonists, we identified novel and important in vitro SARs for the AR-binding affinity and agonist activity (30, 31), including a para-nitro group in the A-ring, a trifluoromethyl group linked to the chiral carbon (R-isomer), a thio-ether linkage, and a halo or para-N-alkylamido group in the B-ring. When these compounds were tested in vivo, however, no pharmacologic activity was observed owing to their unfavorable pharmacokinetic properties (32). Further structural modification was made to overcome this problem by changing the thio-ether linkage to an ether bridge, which resulted in the successful discovery of the first member, S-4, of a new series of SARMs (33, 34). As shown in Figure 3⇓, S- 4 acted as a full AR agonist in the levator ani muscle, as indicated its ability to fully maintain the muscle to that of control level. However, S-4 acted as a partial agonist in the prostate (Emax = 35% of control values), indicating that S-4 was potent and efficacious in anabolic tissues but not in androgenic tissues. In contrast, castrated rats treated with testosterone propionate had near identical, and nonselective growth of the prostate and levator ani muscle as compared to control animals. Moreover, a variety of structural modifications of known SARMs were made to further explore SARs of nonsteroidal AR ligands (Figure 2⇓) and discover novel SARMs having efficacious and potent in vivo pharmacologic activity and favorable pharmacokinetic properties. First, we replaced the para-acetamido group of S-4 with different electron-withdrawing groups, such as halogen, cyano, or nitro groups (type I). Second, we introduced multiple substituents into the aromatic B-ring (type II). Third, we incorporated different electron-withdrawing groups at the ortho, meta and para positions of the A-ring or structurally modified the A-ring to different heterocyclic ring systems (type III). These structural modifications significantly altered AR binding affinity, in vitro functional activity, in vivo pharmacologic activity, and pharmacokinetic properties.
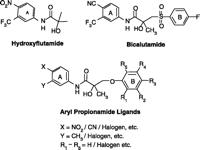
Chemical structures of hydroxyflutamide, bicalutamide, aryl proprionamide ligands.
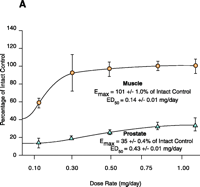
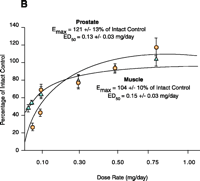
Tissue-selective pharmacologic activity of S-4. A. Male rats were castrated and treated for 14 consecutive days with S-4. S-4 potently stimulates muscle growth, but is unable to maintain prostate size. Adapted from J. Pharmacol. Exp. Therap.304,1334 (2005). B. Testosterone stimulates muscle and prostate growth to the same extent.
For type I SARMs, in vitro AR binding affinity decreased as the size of the halogen atom increased and/or electronegativity decreased (35). In contrast, a chloro-substituted SARM (i.e., S-9) demonstrated the highest in vivo pharmacologic activity. Further pharmacokinetics studies revealed that the terminal half-life of these SARMs ranged from 4.1 to 14.7 hours in the rat and increased as the size of the halogen atom increased. These studies suggested that high efficacy and potency of SARMs should be predicted by two factors, namely high binding AR binding affinity (i.e., Ki < 10 nM) and low in vivo clearance (35). For type II SARMs, the para and meta positions of the B-ring are the optimum positions to introduce small size electron-withdrawing moieties, such as fluoro, chloro, nitro, or cyano groups. The incorporation of a meta-fluoro group in the B-ring of S-9 (to become the novel SARM C-6) resulted in further improved pharmacologic activity in the rat and potential feasibility for hormonal male contraception (36). When the aromatic B-ring was occupied by more than two substituents, the size of the substituents were critical. Partial agonists that were identified using an in vitro functional assay showed no activity in castrated animals. Unlike the aromatic B-ring, the A-ring of our SARM pharmacophore is more restricted in terms of possible structural modifications. Heterocyclic A-ring derivatives failed to retain AR binding affinity (i.e., Ki > 700 nM), which probably arose from steric hinderance upon binding with the AR. For type III SARMs, compounds with a para-cyano group and a meta-halogen group in the A ring maintained high AR binding affinity and in vivo pharmacologic activity in the rat (37).
Pharmacokinetics and Metabolism of SARMs
A series of AR ligands (e.g., acetothiolutamide) studied by Yin et al. exhibited similar AR binding affinity as that of testosterone and high in vitro functional activity; however, these compounds were inactive in the rat owing to rapid hepatic elimination (t1/2 = 26 min) (32). Three major metabolism pathways of acetothiolutamide were identified, including oxidation of the thio-ether linkage, hydrolysis of the amide bond linked to the B-ring, and sulfate conjugation. Replacing the thio-ether linkage of acetothiolutamide significantly decreased the rate of hepatic metabolism and permitted identification of the first member of this series of SARMs, S-4. In the rat, S-4 exhibited linear pharmacokinetics within the pharmacologic dose range after iv administration (38). The lack of parent drug in the urine suggests that S-4 is extensively metabolized. Both in vitro and in vivo metabolism studies showed that S-4 is deacetylated in rats, dogs, and humans (39). The average terminal half-life of S-4 in the rat and dog was approximately four hours. After oral dosing, S-4 was rapidly absorbed and completely bioavailable. We replaced the acetamido group of S-4 with different halogen groups to examine its role in pharmacologic activity. As discussed in the SAR section, these structural modifications significantly altered the in vivo pharmacologic activity of these SARM by changing their AR binding affinity and pharmacokinetic properties. It is worth noting that SARMs with high AR binding affinity (i.e., Ki < 10 nM) exerted nearly identical in vivo pharmacologic activity when normalized to the same level of drug exposure. These results suggested that the in vivo pharmacologic activity of SARMs with high AR binding affinity was largely governed by in vivo drug exposure, whereas high in vivo activity might be difficult to achieve with SARMs of lesser AR binding affinity (i.e., Ki > 10 nM) even when sufficient in vivo drug exposure is achieved (35). Furthermore, we found that the para-nitro group in the A-ring of our SARMs underwent nitro reduction and produced inactive metabolites in the rat (40, 41). Replacing the nitro group with a nonreducible electron-withdrawing group (i.e., a cyano group) could possibly improve the in vivo stability of SARMs. Although the AR binding affinity of cyano-substituted AR ligands is slightly lower than their nitro-substituted counterparts (30, 31), the overall contribution of the cyano group to the in vivo pharmacologic activity was positive. For instance, S-22 bears a cyano group at the para position in both the A-ring and B-ring and was identified as the most potent and efficacious aryl-propionamide SARM that we have observed to date with favorable pharmacokinetic properties (35).
Crystallography and Molecular Modeling of SARMs
To facilitate the rational design of novel and more potent SARMs, three-dimensional models for the human AR ligand binding domain (LBD) bound to testosterone have been developed based on the crystal structure of the highly homologous human progesterone receptor LBD (42). Moreover, the binding modes of several hydroxyfutamide- derived AR ligands were investigated using flexible docking with FlexX, a computer program for predicting protein-ligand interactions. In this work, Marhefka et al. proposed that a unique unoccupied subpocket existed within the AR binding pocket, which may be valuable for ligand optimization of nonsteroidal AR ligands and discovery of novel SARMs. Recently, we examined the threedimensional quantitative structure-activity relationship (QSAR) of a group of endogenous androgens and nonsteroidal AR ligands for the AR using comparative molecular field analysis (CoMFA) (43). In this work, the homology model of the AR developed by Marhefka et al. was used as a scaffold. The integrated homology modeling and CoMFA studies identified key amino acids thought to directly interact with our SARMs. According to these studies, the B-ring was positioned in a subpocket bordered by Met780, Cys784, and Met787.
More recently, the crystal structure of the AR mutant W741L LBD bound to R-bicalutamide at 1.8 Å resolution was solved (44). This mutation confers agonist activity to bicalutamide and may facilitate understanding of the binding modes of bicalutamidederived nonsteroidal AR ligands. Positions of AR residues and the majority of ligand binding plane were similar between the mutant AR W741L-R-bicalutamide LBD complex and wild-type AR-DHT LBD complex (45). However, the absence of the W741 side-chain in the mutant AR W741L allows the B-ring of R-bicalutamide to be accommodated within a region not occupied by DHT and to make direct contract with residues of helix H12. It was proposed by Bohl et al. that the binding modes of AR bound with the aryl-propionamide SARM pharmacophore would be similar to that observed in AR mutant W741L LBD bound to R-bicalutamide. Ongoing crystallography studies in our laboratory focus on the binding modes of SARMs and R-bicalutamide in the wild-type AR LBD and full length AR. Molecular modeling based on the crystal structure of AR–SARM LBD complex should be more useful and accurate than using homology AR models in the design of novel AR ligands and prediction of binding affinity and/or functional activity of novel nonsteroidal AR ligands. In summary, in vitro and in vivo SAR studies show that the aromatic B-ring of the aryl-propionamide SARM pharmacophore is amenable to structural modifications and critical for pharmacologic activity. Although the majority of aryl-propionamide derivatives demonstrated high oral bioavailability in the rat, the other pharmacokinetic properties (e.g., volume of distribution and clearance) varied significantly in vivo. In vitro AR binding affinity, intrinsic activity of the ligand, and in vivo drug exposure contribute to the overall in vivo potency and efficacy of SARMs. Molecular modeling of nonsteroidal AR ligands is used constantly, in conjunction with pharmacology, pharmacodynamics, pharmacokinetics, and metabolism, to examine and predict the best structural properties. Enhanced understanding of the molecular interactions between nonsteroidal ligand and the AR has advanced significantly in the last five years and, with time, will lead to further structural optimization and discovery and development of SARMs.
Therapeutic Promise of SARMs
General
SARMs are currently in the early stages of development, meaning that only animal data is available and that limited endpoint markers have been studied. Nevertheless, the development of SARMs for clinical uses is promising based on preclinical data. Owing to their selectively high anabolic activity, SARMs could be used for prevention or treatment of many diseases, including muscle wasting, osteoporosis, frailty, or other conditions associated with aging or androgen deficiency––without unwanted side effects associated with testosterone. In addition, SARMs that act as partial agonists in the prostate could be used to prevent or treat BPH. Likewise, SARMs might be used for hormonal male contraception. In contrast to the overall pharmacologic activity of testosterone, which arises from testosterone, DHT, and estradiol, SARMs lack estrogenic-like activity and do not demonstrate amplified activity in the prostate. Estrogen is important in bone development and its effects might be related to cognitive function, libido, and cardiovascular function in the male. It is still unknown whether SARMs alone will cover the full spectrum of beneficial effects provided by testosterone replacement or not. Near-term phase II and III clinical trials of SARMs should provide insight to their potential therapeutic use.
Treatment of Muscle Wasting
Testosterone replacement in young hypogonadal men at a physiologic dose is associated with changes in body composition (i.e., gain of lean mass and loss of fat mass) and increase in muscle protein synthesis (12, 46). Moreover, administration of testosterone at a physiologic dose increases maximal voluntary strength in young hypogonadal men (46). Administration of a supraphysiologic dose of testosterone (about six times the dose needed to achieve normal serum concentrations) to healthy normal men increases fat-free mass to a similar extent as resistance-exercise training. The combinational therapy of resistance exercise and a supraphysiologic dose of testosterone showed additive effects on fat-free mass and muscle size and strength. Similarly, administration of a supraphysiologic dose of testosterone is associated with increase in maximal voluntary strength and quadriceps cross-sectional area and volume (47).
The majority of studies to date examined the effects of testosterone replacement in older men. Generally, testosterone replacement in elderly men produced modest increases in muscle mass and strength. Some studies reported modest increases in lean mass (i.e., whole body mass excluding bone and fat mass) (48). Few studies, however, have reported that androgen administration increases grip strength (49); in fact, at least one has shown that it does not (50). Likewise, the effects of testosterone administration on lower-body strength were not significant in several studies (48). Although the effect of testosterone on muscle mass and strength in elderly men has not been consistent or impressive, these results do not suggest that the administration of SARMs would not be beneficial in terms of muscle mass and strength in elderly men. Only lower doses of testosterone were considered for studies in elderly men, owing to the concerns of side effects at higher doses, especially accelerating the risk of prostate cancer. A study showed that administration of testosterone at 125, 300, or 600 mg/week significantly increased muscle mass and strength equally in older and younger men. These actions of androgen in muscle seem to have a dose (and concentration)- dependent relationship (51). In healthy young men, testosterone dose and testosterone concentrations, including total, free, and steady-state concentration, were highly correlated. Also, there were positive correlations between the testosterone dose administered and gains in fat-free mass, leg press strength, and leg power, whereas testosterone dose and fat mass were inversely correlated. Moreover, preoperative administration of supraphysiologic doses of testosterone tended to shorten hospital stay and improve walking and stair climbing in older men undergoing knee replacement surgery. At postoperative day 3, there was a significant improvement in the testosterone treatment group in the ability to stand (52).
It would seem that aromatization of androgens to estrogen is not required for mediating their anabolic effects on muscle. Both testosterone and non-aromatizable androgen (i.e., nandrolone) increased muscle mass and strength, and there was no significant difference between testosterone and nandrolone in the magnitude of increase in muscle (53). Also, males with a dysfunction of estrogen action have normal muscle phenotypes.
SARMs demonstrate strong agonistic activity and an ability to promote growth of the levator ani muscle, maximally to a size significantly greater than that of intact control animals (54). However, the anabolic activity of SARMs in the levator ani muscle does not directly support the contention that SARMs will improve muscle performance. Recently, the effects of S-4 on the mass and strength of skeletal muscle (isolated soleus muscle) in orchidectomized rats were measured (55). S-4 treatment (3 mg/kg and 10 mg/kg) significantly increased the skeletal muscle strength (measured as peak titanic tension) in orchidectomized animals, even though the effect of S-4 in muscle size was not significant. S-4 restored castration- induced losses in lean body mass. Similar changes in muscle size, muscle strength, and lean body mass were also observed in DHT-treated (3 mg/kg) animals. However, DHT (3 mg/kg) also fully restored the androgenic tissue weights, whereas S-4 (3 mg/kg) only returned the prostate and seminal vesicle to 16% and 17%, respectively, of the control levels.
In summary, administration of androgen significantly increases muscle mass and strength in young hypogonadal men (physiologic replacement dose) and eugonadal men (supraphysiologic dose). In elderly men, testosterone effects on muscle mass and strength have not been consistent or impressive, possibly due to the low dosages used in clinical trials. The high correlation between dose (and concentration) and the anabolic actions of androgen in muscle suggests that androgen administration of higher doses in elderly men may significantly increase muscle mass and strength. The aromatization of androgens to estrogen is not required for mediating their anabolic effects on the muscle, suggesting that SARMs can also increase muscle mass and strength. In orchidectomized animals, S-4 showed strong anabolic effects in skeletal muscle without affecting the androgenic tissues. This evidence strongly supports the great potential of SARMs as anabolic agents to treat muscle wasting, improve muscle performance in the frail, and shorten rehabilitation time after surgery.
Prevention and Treatment of Osteoporosis
Numerous lines of evidence indicate that androgens are important in bone, and that SARMs may represent a novel approach to treatment of osteoporosis. Reduction of androgen concentration correlates well with bone resorption markers in men admitted for hip fracture, suggesting that loss of androgen may contribute to bone loss and increased risk of fracture (56). Loss of androgen by surgical castration increases the cumulative incidence of fracture in prostate cancer patients (57). Androgen replacement prevents bone loss and increases BMD in hypogonadal men (58); however, the effect of androgen replacement on fracture risk remains unknown. Women with hirsutism and polycystic ovary syndrome, who have excess endogenous androgen, have increased BMD compared with normal young women (59). Estrogen and androgen therapy increases BMD to a greater degree than does estrogen therapy alone (60).
An important issue from the perspective of developing SARMs as anabolic agents for bone is whether aromatization of androgen is necessary for anabolic action. In humans, androgen-insensitive males that have inactivating AR mutations are frequently osteopenic (61). In experimental animals, non-aromatizable androgens significantly prevent osteopenia that results from orchidectomy (62). DHT also prevents bone loss after ovariectomy in female rats (63), and flutamide (an AR antagonist) induces osteopenia even in estrogenreplete female rats with intact ovaries (64). In addition to this indirect evidence from studies of steroidal androgens, several studies of SARMs have already demonstrated their potential as a treatment for osteoporosis. SARMs significantly increased BMD and bone strength in orchidectomized and ovariectomized rats (65–67). Hanada et al. reported that a SARM (i.e., S-40503) induced a significant increase in femoral BMD in a dose-dependent manner in a hypogonadal model, with lesser activities in androgenic organs. Rosen and Negro- Vilar also reported that a SARM (i.e., LGD2226) prevents bone loss observed in orchidectomized animals. In ovariectomized rats, SARMs demonstrated a significant elevation in femoral BMD (S- 40503) and mechanical strength of femoral bone (S-4 and S-40503).
The majority of clinically available therapies for osteoporosis are bone resorption inhibitors (such as bisphosphonates, estrogen, SERMs, and calcitonin), which are not sufficient to restore bone mass for patients who have already lost a significant amount of bone. Intermittent parathyroid hormone (PTH) treatment is the only clinically available option to promote bone formation; however, PTH treatment for osteoporosis is very limited because of side effects and possible association with osteosarcoma. The lack of satisfactory treatments has piqued interest in use of SARM as a treatment of osteoporosis. That interest still remains appropriate because several lines of evidence support the anabolic activities of SARMs, especially at periosteal cortical bone. Androgen seems to be responsible for obvious sexual differences in skeletal morphologies after puberty. The skeletal sizes of men––including radial width and cortical thickness of long bone, femoral neck cross-sectional area, and vertebral cross-sectional area––are larger than those in women (68). In humans and rodents, genetic males with complete androgen insensitivity gain a bone mass more typical of genetic females, suggesting the anabolic action of androgen is critical for the accumulation of significant muscle mass (68). Turner et al. showed that DHT increases bone formation in the periosteal surface of cortical bone, whereas estrogens depress it (69). Even though volumetric BMD [i.e., the amount of mineral per volume of bone (g/cm3)] is very similar in adult men and women, the larger diameter and cortical thickness of the long bones in men offers great advantage to their mechanical strength, explaining the lower incidence of fracture of male bones compared with that of female bones (70). Hanada et al. provided direct evidence for the anabolic action of SARMs in bone––the only published evidence to date (65). In this study, a SARM, S-40503, elevated the periosteal mineral apposition rate in cortical bone area of ovariectomized rats, demonstrating the bone-forming activity of SARM in the periosteal surface of cortical bone. In orchidectomized rats, S-40503 treatment significantly increased cortical BMD in femoral and tibial bones, but did not affect the cancellous BMD in both bones, suggesting that SARMs significantly promote BMD mainly by anabolic action, rather than by anti-resorptive action. It is worth noting that S-40503 treatment enhanced cortical BMD to a similar extent as did estrogen in an immobilized, orchidectomized model of osteoporosis. In fact, S-40503 increased cortical BMD significantly higher than intact and estrogen treatment groups in orchidectomized rat. This observation suggests that a SARM might increase BMD by direct anabolic action and indirect action through muscle stimulation, even though this comparison was performed in different bones.
In conclusion, SARMs are still in the early stages of drug development and knowledge of their activity in the skeleton remains sparse; nonetheless, SARMs have great potential for treatment of osteoporosis. First, their unique tissue selectivity might make for the beneficial usage of AR ligands as a treatment of osteoporosis. SARMs can minimize undesirable side effects, resulting from stimulation of androgenic organs and cross-reactivity of androgens and their metabolites. Moreover, the anabolic effect of SARMs in bone could likely be promoted by higher dosage regimen than conventional dosage for steroidal androgen, because conventional steroidal androgen dosages are restricted by side effects. Second, SARMs promote bone formation, rather than reduce resorptive action, which suggest that SARM can restore bone mass even for severe osteoporosis as well as preventive and early stage osteoporosis. Combination therapy with other anti-resorptive agents might synergistically increase bone mass and strength. Finally, the anabolic effects of androgen on muscle are beneficial for increasing bone mass and reducing fracture risk. The pharmacokinetic advantages, selectivity, and dual activity of SARMs in muscle and bone suggest that they may indeed become an important new addition to the armamentarium of drugs to treat osteoporosis.
Hormonal Male Contraception
Unlike female contraceptive measures, effective male contraception is restricted to physical methods, namely condoms and vasectomy. A variety of attempts have been made to produce pharmacologic, effective, reversible and side effect–free contraceptive methods for the male. Among them, only hormonally-based contraception has reached the stage of clinical development. Theoretically, inhibition of one or more hormones involved in the hypothalamus-pituitary-testis axis (Figure 1⇑) will suppress spermatogenesis. GnRH secretion can be inhibited either by exogenous androgens via negative feedback or by GnRH agonists. At the pituitary level, the stimulatory effect of GnRH can be blocked either by competitive binding of GnRH antagonist with GnRH receptors or by a GnRH antibody that binds GnRH before it interacts with its receptor. Progestins and exogenous androgens inhibit secretion of LH and FSH, while inhibin can specifically decrease the amount of secreted FSH. Additionally, the effects of FSH on Sertoli cells can be blocked using either FSH immunization or FSH antagonists; however, the only clinically-proven effective methods for men are androgen alone and androgen combined with either progestins or GnRH antagonists [for review, see (3)]. The use of FSH immunization, FSH antagonists, and inhibin for hormonal male contraception is in its infancy. For successful male-specific contraception, the production of LH, FSH, and consequently, intratesticular testosterone need to be maximally suppressed, while the physiologic needs of peripheral androgens are required to be supplemented by exogenous means.
In the 1990s, two large international studies sponsored by the World Health Organization (WHO) (71, 72) tested testosterone enanthate (TE), by im injection, as a male contraceptive at a dose of 200 mg/week. Subjects did not use any other contraceptive for one year once their sperm concentrations had fallen below the set threshold. The overall pregnancy rate was 1.4% when jointly considering the azoospermia and oligozoospermia groups (0.1 to 3.0 x 106 sperm/mL) and 8.1% in the oligozoospermia group alone. Although these WHO studies provided proof-of-concept support for using testosterone for hormonal male contraception, they did not offer a practicable method. Recently, the efficacy of male contraception using a longer-acting androgen (i.e., testosterone undecanoate, TU) was studied in China. Monthly injection of TU at a dose of 500 mg for 12 months demonstrated a 97% success rate in achieving azoospermia and oligozoospermia and an overall failure rate in preventing pregnancy of 5.2 per 100 person-years (73). The main disadvantages of using a testosterone-alone regimen are the inconvenient dosing method, pain of injection, slow onset (i.e., four months) and steroid androgen–related side effects, including increased body weight, acne, and changes in serum lipoproteins and possible effects in the prostate. Additionally, azoospermia was only observed in 50 to 70% of Caucasian men, whereas a higher rate (91%) of azoospermia was achieved in an East Asian population using testosterone alone. Possible explanations of this heterogeneity in the spermatogenic response include the sensitivity of hypothalamus-pituitary-testis axis to the negative feedback signal from testosterone (74), intratesticular 5α-reductase activity (75), pretreatment hormonal status (76, 77), and polymorphisms in the common polyglutamine stretch (CAG repeats) length in exon 1 of the AR gene (78).
To increase the response rate of hormonal male contraception in Caucasians and to avoid those disadvantages caused by supraphysiologic doses of testosterone, other gonadotropin-suppression substances (e.g., progestins and GnRH antagonists) were included and used as combination regimens. The widespread use of GnRH antagonists for hormonal male contraception is limited because these proteins are expensive to synthesize and difficult to deliver. On the contrary, progestins have been used as a key component of female contraception for decades. The administration of progestins alone in men results in faster and more potent gonadotropin suppression (79); however, nearly complete depletion of endogenous testosterone leads to loss of libido. Therefore, a physiological dose of testosterone was always included during most clinical trials. Additionally, testosterone and progestins work synergistically in suppression of gonadotropins. The currently available progestins include cyproterone acetate (CPA), depot medroxyprogesterone acetate (DMPA), norethisterone enanthate (NETE), levonorgestrel (LNG), desogestrel (DSG), etonogestrel (ENG), and dienogest (DNG). Except for DMPA, all other progestins are orally active compounds, which are good candidates for inclusion in a male contraceptive “pill.” Clinical trials of hormonal male contraception are aimed at finding the best androgen-progestin combination and the minimum effective dose of these compounds [for recent reviews, see (3, 80)]. Many studies based on weekly or biweekly injection of TE in combination with oral or depot forms of progestins have shown profound sperm suppression and tolerable side effects. Unfortunately, the major limitations of these studies to date have been the small population involved in each study and unpractical dosing regimens. Several novel regimens were studied recently, including a self-applied regimen using a testosterone transdermal patch plus DSG at doses of 75, 150, and 300 μg/day (81). Owing to the failure of delivering sufficient androgen, the azoospermia rate was significantly lower than those therapies consisting of TE- or T-pellets combined with the same dose of DSG. More recently, a depot testosterone/DMPA combination regimen was investigated in healthy men (82). Although this dosing regimen demonstrated high efficacy with little-to-no pregnancies observed, approximately one-half of the volunteers discontinued the study for various reasons. Additionally, the administration routes used in this study, implantation and im, were inconvenient despite the fact that the dosing frequency was relatively low. TU alone demonstrated promising efficacy, therefore, more recent studies have focused on long-acting androgens combined with potent progestins, such as TU + NETE and testosterone decanoate (TD) + ENG. In the most recent multicenter clinical trial involving 112 healthy men, treatment with 300 μg ENG daily plus 400 mg TD every four or every six weeks for forty-eight weeks achieved 95.3% and 82.5% azoospermia in the respective treatment groups (83). To date, progestins (e.g., norethisterone, desogestrel, etonogestrel, and DMPA) combined with long-acting testosterone esters (e.g., TU and TD) appear to represent the most promising approach to hormonal male contraception.
No major toxicological effects were reported in most hormonal male contraception clinical trials involving young eugonadal subjects. Besides the inconvenient administration routes, steroid-related side effects are the major limitations of testosterone-based male contraception and include decreases in HDL cholesterol, increases in hematologic parameters such as hemoglobin and hematocrit, increase in body weight, gynecomastia, and acne. Although lower doses of testosterone were effectively used in testosterone-progestin combination regimens, similar but minor side effects were reported. Additional concern of long-term treatment with testosterone is the potential risk of testosterone in the prostate and cardiovascular system, which needs to be evaluated by large and long-term prospective studies in the future.
To prevent potential adverse effect of androgen treatment on the prostate, tissue selective steroidal androgens, such as 7α-methyl- 19-nortestosterone (MENT), were recently investigated for hormonal male contraception. MENT is an aromatizable but non-5α-reducible steroidal androgen, which demonstrated four- and twelve-times more potency than testosterone in the prostate and levator ani muscle, respectively, in castrated male rats and maintained sexual behavior in animals and in hypogonadal men. The antireproductive activity of MENT was recently studied in animals and men. High efficacy (with 100% infertile rate) was achieved using MENT alone or in combination with estradiol as hormonal contraception in adult male bonnet monkeys (84). Most recently, MENT’s reversible effects and its usefulness as a single-regimen hormone-driven male contraceptive have been investigated in early-phase clinical trials (85). MENT acetate implants of differing doses were inserted subdermally at a target delivery dose rate of 400 μg/day for twelve months. In the highest dose group, azoospermia was achieved in 72% of subjects, and 9% exhibited oligospermia. One was oligozoospermic and the remaining two were nonresponders. Previous pharmacologic studies in the monkey revealed that MENT maintained normal size of the prostate at the minimum dose required to suppress LH (86). Consistently, no significant difference in the size of prostate was observed before and after treatment in all drug treatment groups. Although the effective dose of MENT is much lower than that required in testosterone-based male contraception regimens, typical steroid-related side effects were similar to those reported in testosterone- based clinical trials. Additional pharmacokinetic studies showed that the total body clearance of MENT in both monkey and men was much greater than that of testosterone. The long-term action of MENT after a single implantation in suppression of spermatogenesis makes it a promising candidate for hormonal contraception either alone or in the combination with progestins. Nevertheless, unfavorable pharmacokinetic properties, the requirement of parenteral administration, and steroidal-related side effects are major limitations of MENT.
Major limitations associated with steroidal androgens can be largely overcome by novel SARMs. An aryl-propionamide SARM, C-6, exhibited tissue selectivity in castrated male rats with higher anabolic activity (ED50, levator ani muscle = 0.68 mg/kg/day) than androgen activity (ED50, prostate = 3.1 mg/kg/day) (36). In the castrated male rats, levator ani muscle mass was maintained at a level similar to levator ani muscles from intact controls by C-6 at a dose of 1.2 mg/kg/day, whereas the mass of the prostate was only partially maintained (50% of control). At this dose, the elevated concentrations of LH and FSH in castrated animals were fully or partially suppressed, respectively. A pilot study of C-6 in intact male rats shows that this SARM significantly inhibits spermatogenesis at a single high dose of 4 mg/kg/day and maintains its tissue-selective pharmacologic activity in the muscle and prostate. Studies of spermatogenesis over a broad pharmacologic dose range of C-6 are needed to fully delineate its effects on spermatogenesis and its feasibility for male contraception. Additional pharmacokinetic studies of C-6 in rats have indicated that it is rapidly and highly (76%) absorbed after oral doses and is cleared slowly from the body (0.72 mL/min/kg). Although the effects of C-6 on hepatic enzymes––such as the aspartate aminotransferase AST-SGOT and the alanine transaminase ALT-SGP––and serum lipids were not evaluated in this study, previous pharmacology studies demonstrated the absence of significant changes in hepatic function and lipid profiles using other SARMs with same pharmacophore in the rat (33). Importantly, the minimum dose of C-6 required for LH suppression and full maintenance of levator ani muscle weight are similar, but at least two-fold less than that needed to fully maintain the prostate. Additionally, the favorable pharmacokinetic properties and oral bioavailability of SARMs make them amenable for an oral daily dose. Furthermore, SARM-treatment appears to be free from unwanted side effects related to steroidal androgens. Taken together, these results indicate that some SARMs (e.g., C-6) are promising candidates for development as a component in a “male pill.”
Treatment and Prevention of Benign Prostate Hyperplasia (BPH)
BPH is a common disease associated with both aging and androgens. The principle intraprostatic androgen is DHT, which is converted from testosterone mainly by type 2 5α-reductase. Without treatment, long-standing BPH potentially leads to recurrent bladder infection, bladder calculi, acute urinary retention, and possibly the necessity of prostate surgery (87). Lower urinary tract symptoms (LUTS), commonly associated with symptomatic BPH, are caused by mechanical blockage of DHT-dependent hyperplasia and reduction of the urethral lumen diameter arising from contraction of smooth muscles under an increased α-adrenergic tone (88). Current drug treatments for BPH include androgen deprivation, phytotherapy, 5α-reductase inhibitors, and α1-blockers. For patients with severe BPH, surgical treatments provide the most rapid and best relief of symptoms.
Androgen deprivation using GnRH analogs, such as leuprorelin and goserelin, suppresses the production of gonadotropins and consequently testosterone synthesis, which significantly decreases the size of the prostate. However, severe side-effects related to androgen deficiency (e.g., loss of libido, hot flashes, and impotence) are inevitable (88). An extract from the American dwarf palm (Serenoa repens) is the most common phytotherapy for BPH. In fact, plant extracts are the first line of treatment for prostate enlargement and associated LUTS in Europe. Permixon, the n-hexane liposterolic extract of S. repens, is currently manufactured in France and marketed in Europe. Efficacy studies of S. repens extract for BPH treatment suggest that it provides mild to moderate improvement in urinary symptoms and urinary flow comparable to that achieved by finasteride, but with less unwanted side-effects (89). Possible mechanisms of S. repens extract efficacy in BPH were recently reviewed and proposed, including antiandrogenic action, anti-inflammatory effects, induction of apoptosis, and antiproliferative activity (90). Understanding the mechanism of S. repens extract in BPH will likely provide a rational basis for the design of novel compounds with specific target(s).
Based on the pathologic mechanisms of BPH, the clinically preferred medical treatment for BPH is either a 5α-reductase inhibitor or an α1-adrenergic blocker. Finasteride was the first available type 2 5α-reductase inhibitor. It reduces prostate size by blocking the conversion of testosterone to DHT in the prostate, thus inducing epithelial atrophy. Long-term finasteride therapy for BPH is effective and safe as proved by multiple efficacy and safety studies. Clinical trials have shown that finasteride consistently decreases prostate volume approximately by 19 to 27%, significantly improves symptoms, and decreases the risk of urinary retention and the likelihood of BPHrelated surgeries in men with symptomatic BPH at a daily dose of 5 mg (91, 92). Moreover, finasteride causes an average reduction of serum prostate specific antigen (PSA) levels of about 50%. However, men with small prostate size at baseline showed less response to finasteride treatment than those with large prostate size. Compared with placebo, ejaculatory dysfunction related to decreased prostatic secretions and decreased libido and/or impotence are commonly associated with finasteride therapy (91). Owing to the inhibition of intraprostatic 5α-reductase activity, plasma testosterone levels increase during finasteride therapy. Consequently, more estradiol may be produced from the increased amount of testosterone via aromatization. In turn, it raises a theoretical concern of stimulating latent prostate cancer by finasteride treatment for BPH. Recently, a landmark Prostate Cancer Prevention Trial (PCPT) (93), involving 18,882 men (≥55 yr), found that finasteride provided a 25% reduction in prevalence of prostate cancer over seven years. Surprisingly, tumors with high Gleason grade (e.g., ≥7, a moderate-to-high grade of prostate cancer) occurred more frequently in the finasteride group than in the placebo group. Less urinary symptoms but more sexual side-effects were observed in the finasteride-treated group than in men receiving placebo. Therefore, the overall risk–benefit of using finasteride for prostate prevention needs to be carefully evaluated in the future.
More recently, a dual inhibitor of 5α-reductase types 1 and 2 was introduced for clinical use of BPH. In contrast to finasteride, which decreases plasma and intraprostatic DHT levels by about 70% and 85%, respectively, dutasteride shows a greater than 90% reduction in plasma concentrations of DHT. Dutasteride at an oral dose of 0.5 mg/day significantly decreased: 1) prostate size by 25%, 2) symptoms of obstruction, and 3) the risk of acute urinary retention or surgical intervention by 48% in a two-year clinical trial (94). A follow-up safety and efficacy study (95) showed no additional side-effects were observed after four-year treatment. There are no published studies directly comparing the efficacy of dutasteride and finasteride; however, comparison of end point markers from separate dutasteride and finasteride studies suggests that they exhibit similar efficacy and side-effect profiles. Additional common adverse effects associated with dutasteride were ear-nose-throat infections, musculoskeletal pain, and upper respiratory infections (94). The potential benefit offered by the more potent suppression of DHT with dutasteride is under investigation.
The α1-adrenergic receptor antagonists (i.e., α1-blockers) improve urinary symptoms by acting on the dynamic component associated with an α1-adrenergic receptor–dependent increase in smooth muscle tone. α1-Adrenergic receptors also play an essential role in regulating blood pressure, thus, developing α1-blockers with tissue-selective action for BPH is the main obstacle. Significant progress has been made in the past decade as evidenced by the discovery and development of several generations of compounds with increased efficacy and safety, namely from nonselective a-blocker (e.g., phenoxybenzamine) to selective α1-blockers (e.g., prazosin, doxazosin, and terazosin) and then to uroselective α1-blockers (e.g., tamsulosin and alfuzosin) [for review, see review (94)]. Combination therapy with a 5α-reductase inhibitor and a selective α1-blocker showed better efficacy than monotherapy with either component (96), indicating that such combination therapy is a promising regimen for the treatment of BPH.
SARMs acting as a partial agonist in the prostate but a full agonist in the muscle may provide a novel therapeutic approach for the treatment of BPH. The pharmacologic activity of the SARM S-1, hydroxyflutamide (antiandrogen), and finasteride (5α-reductase inhibitor) in intact male rats was recently reported (97). Additionally, S-1 (5, 10, and 25 mg/kg) selectively decreased the prostate weight and was equal in efficacy to finasteride (5 mg/kg). At the end of the nine-day treatment, no significant changes in levator ani muscle weight, plasma levels of testosterone, or FSH were observed in the SARM-treated group. Nonetheless, finasteride significantly increased testosterone concentrations in intact male rats. The SARM also showed very weak inhibition of human 5α-reductase enzymes, both types 1 and 2, suggesting a different mechanism in suppressing prostate size other than that of finasteride. These studies indicate that SARMs may be feasible for the treatment of BPH either as a single or combination therapy in the future.
Future Directions
Results from in vitro and in vivo animals studies suggest that the therapeutic promise of SARMs as treatment for muscle wasting, osteoporosis, hormonal male contraception, and BPH may be realized in the not so distant future. Demonstrated advantages, including tissue selectivity, favorable pharmacokinetic properties, AR specificity, and lack of steroidal-related side effects, clearly distinguish these drugs from their steroidal predecessors and open the door for expanded clinical use of androgens. The first potential clinical application of SARMs is most likely to be the treatment of muscle wasting. The rapid and profound improvement upon treatment with testosterone makes muscle-wasting an ideal and relevant target for early clinical trials and proof-of-concept studies with SARMs. As the molecular mechanisms of action of SARMs on target tissues become more fully understood, the discovery of novel SARMs and expansion into broader therapeutic applications will be more feasible. Currently, research on SARMs is in its early stages, namely preclinical discovery and the early phase of clinical development. Phase II studies planned in the next two to three years, however, should reveal the true promise of this exciting new therapeutic class of drugs. It will take years of efforts to deliver SARMs from the laboratory bench to patients. It is to be hoped that ideal SARMs with all of the beneficial pharmacologic activity of androgens without the unwanted side effects will provide individual patients with various androgen-dependent conditions a significantly improved quality of life.
Acknowledgments
Our SARM studies reported herein were supported by grants from NIH (R01 DK59800), GTx Inc., and a Presidential Fellowship, The Ohio State University.
- © American Society for Pharmacology and Experimental Theraputics 2005
References

Jiyun Chen, PhD, (left) was a graduate student and Presidential Fellow in Dr. Dalton’s laboratory at The Ohio State University. Her dissertation research focused on the structure-activity relationships for nonsteroidal selective androgen receptor modulators and their potential application to hormonal male contraception. She is currently a research scientist at Amgen, Inc., Thousand Oaks, CA. E-mail: chenjiyun{at}yahoo.com.
Juhyun Kim (right) is a graduate (BS and MS) of Chosun University (South Korea) and current graduate student in Dr. Dalton’s laboratory at The Ohio State University. Her dissertation research focuses on the pharmacokinetics and metabolism of nonsteroidal selective androgen receptor modulators and their potential application to osteoporosis. E-mail: kim.1452{at}osu.edu
James T. Dalton, PhD, (center) is Professor of Pharmaceutics in the College of Pharmacy at The Ohio State University. He is currently on leave of absence from the university and serving as Vice-President of Preclinical Research and Development at GTx, Inc., Memphis, TN (www.gtxinc.com), a men’s health biotech company leading clinical development of nonsteroidal selective androgen receptor modulators. Dr. Dalton is a co-inventor on over 50 US and international patents and patent applications on SARMs. His research interests include the molecular, preclinical, and clinical pharmacology of novel drugs, with an emphasis on selective nuclear receptor modulators and anticancer agents. E-mail: jdalton{at}gtxinc.com or dalton.1{at}osu.edu, fax: 614-292-7766.

