The Neuronal Death Protein Par-4 Mediates Dopaminergic Synaptic Plasticity
The concept of signaling cascades and molecular interactions whose only function is to kill cells is fading in the face of accumulating evidence that such “cell death pathways” also mediate adaptive physiological responses of cells to changing environments. The identification of key proteins involved in a form of programmed cell death called apoptosis has had a major impact on biology and medicine, with the fields of cancer and neurological disease being two prominent examples. The goal of cancer specialists is to selectively kill abnormal cells, whereas neuroscientists aim to prevent the dysfunction and death of nerve cells. A small but growing list of proteins involved in the immortality or killing of cancer cells have been implicated in physiological functions in the nervous system including neurogenesis, neurite outgrowth, and synaptic plasticity (1). The latest addition to this list is prostate apoptosis response-4 (Par-4), a protein discovered in a genetic screen to identify genes whose expression is increased in prostate cancer cells undergoing apoptosis (2). Subsequent studies suggested a pivotal role for Par-4 in the deaths of neurons that occur in experimental models of Alzheimer Disease, Parkinson Disease, amyotrophic lateral sclerosis, and stroke (3–6). Now, in an elegant and comprehensive series of experiments, Park et al. provide evidence that Par-4 plays an unexpected role in regulating neurotransmission at synapses in the striatum that employ the neurotransmitter dopamine (7). Remarkably, their findings show that impairment of Par-4 function at dopaminergic synapses can result in a depression-like syndrome in mice. Thus, in one fell swoop the authors raise Par-4 from a killer waiting for an opportunity to destroy neurons, to a hero ensuring the smooth operation of the neurons it serves.
The binding of Par-4 to other proteins is mediated by its leucine zipper (LZ) domain, which is essential for its pro-apoptotic function (3). The amount of Par-4 increases rapidly in neurons subjected to several different triggers of apoptosis including trophic factor deprivation, oxidative stress, and excitotoxins (3–6). Par-4 may promote apoptosis by binding to atypical forms of protein kinase C (8), Dlk [death-associated protein (DAP)-like kinase]/ZIP (zipper-interacting protein) kinase (9), and/or the Wilm’s tumor suppressor protein (10). By inhibiting cell survival pathways that involve, for example, the transcription factor NF-κB and increased expression of Bcl-2, Par-4 can block the ability of cells to protect themselves (11). Par-4 is present in low amounts in neurons throughout the brain under normal conditions and is located in dendrites and synapses where it can mediate local degeneration under conditions of exposure to severe oxidative and excitotoxic insults (12). The findings of Park et al., however, reveal a new and intriguing function of Par-4 at dopaminergic synapses (7). In a yeast two-hybrid screen they identified Par-4 as a protein that interacts with the D2 dopamine receptor receptor (D2DR). The authors then showed that these two proteins interact in neural cells and that they are both expressed in striatal neurons that receive dopaminergic inputs. The interaction is mediated by the LZ domain of Par-4, which binds to a calmodulin-binding domain of the D2DR. Calmodulin displaces Par-4 from D2DR in a calcium-dependent manner, suggesting a potential role for Par-4 in modulating calcium-mediated dopaminergic signaling.
By using RNA interference technology to deplete Par-4 from cultured cells and by studying mice expressing a mutated form of Par-4 lacking the LZ, the authors provide direct evidence that Par-4 normally functions to enhance D2DR signaling and thereby increases the inhibition of dopamine-mediated neurotransmission (7). Indeed, activation of the cAMP/calcium-activated transcriptional regulator CREB (cyclic AMP response element binding protein) was increased in striatal neurons from Par-4–mutant mice (Figure 1⇓). To understand the consequences of an impaired functional interaction between Par-4 and D2DR, the behaviors of mice with impaired Par-4 function were evaluated. The Par-4 mutant mice exhibited abnormal behaviors (i.e., immobilization or “freezing”) in the forced swim and tail suspension tests, which are understood to model a depression-like state in these mice. The impaired D2DR signaling in the Par-4 mutant mice may account for their depression-like behaviors because previous studies have provided evidence for impaired dopaminergic signaling in clinically depressed humans (13). More strongly linked to depression, however, are abnormalities in serotonin and norepinephrine signaling pathways (14). Park et al. (7) did not determine whether there were abnormalities in the serotonergic or noradrenergic systems in the brains of the Par-4 mutant mice, leaving open the possibility that Par-4 modulates these depression-linked signaling pathways. The depression-inducing effect of Par-4 dysfunction might also result from impaired neurogenesis because Par-4 plays an important role in neurogenesis (15), and impaired neurogenesis has been associated with depression (16). In addition, the widely prescribed mood-stabilizing drug valproate increases the expression of Par-4 in the brain of adult mice (17), suggesting a role for Par-4 in the therapeutic actions of valproate.
The findings described above add Par-4 to a growing list of cell death-related proteins that may serve other important functions in the nervous system (Figure 1⇓). Two other examples of proteins that were initially identified as playing pivotal roles in apoptosis and subsequently shown to regulate neurite outgrowth and synaptic plasticity are caspase-3 and Bcl-2. Caspase-3 is one member of a family of cysteine proteases that execute the cell-death process in many different types of cells including neurons (18). Studies of hippocampal neurons have shown that specific glutamate receptor subunits are substrates of caspase-3, and that their cleavage negatively regulates responses to glutamate (19). Biochemical and electrophysiological analyses demonstrated a direct action of caspase-3 on the glutamate receptor subunit GluR1 that results in reduced ion currents through the α-amino-3-hydroxy-5-methylisoxazole-4-propionate (AMPA) receptor (20). More recently, Lynch and coworkers have provided evidence that caspase-1 (also known as interleukin-1β–converting enzyme or ICE) can inhibit long-term potentiation of synaptic transmission in the hippocampus (21), suggesting a role for this caspase in physiological and pathological impairment of learning and memory. Anti-apoptotic proteins such as Bcl-2 and Bcl-xL may prevent apoptosis by stabilizing mitochondrial membranes and by modulating calcium fluxes in mitochondria and the endoplasmic reticulum (22). Microinjection of the anti-apoptotic Bcl-xL protein into the presynaptic terminal of the adult squid stellate ganglion enhanced postsynaptic responses and the rate of recovery from synaptic depression, whereas injection of a pro-apoptotic Bcl-xL cleavage product suppressed postsynaptic responses (23). Mice with targeted disruption of one allele of Bcl-2 exhibit increased anxiety-like behaviors (24), which suggests a role for Bcl-2 in modulating the neural circuits that control anxiety. The specific mechanisms by which Bcl-2 family members modify synaptic plasticity are unknown.
From an evolutionary perspective it is, perhaps, not surprising that the same proteins that control the dramatic and irreversible process of apoptosis also serve important roles in regulating dynamic changes in the function and structure of neural circuits. The proteins (e.g., Par-4, caspases, Bax, p53, etc.) are present in neurites and synapses in low amounts under normal conditions. Intuitively, it is unlikely that these proteins are expressed throughout adult life with the sole function of waiting for a disease or trauma to activate them. The emerging picture, clarified by the findings of Park et al. (7), is that apoptotic proteins serve important roles in the local regulation of structural remodeling of synapses and neurites in response to sublethal activation of “death signaling” pathways including those activated by glutamate, calcium, and oxidative stress (25, 26). Most of the known components of apoptotic biochemical cascades have been shown to be located in synapses and neurites (Table 1⇓). In addition to Bcl-2 family members and caspases these include: death receptors [tumor necrosis factor–α (TNF-α) receptors and Fas] and their downstream transducers, the tumor suppressor protein p53, cytochrome c, and inositol 1,4,5-trisphosphate (IP3) receptors. A better understanding of the functions of apoptotic and antiapoptotic proteins in the contexts of synaptic function, plasticity and disease will likely result in novel therapeutic approaches for a range of neurological conditions that includes not only those involving cell death, but also functional disorders.
Examples of “Apoptotic” Proteins Located in Synapses and Neurites
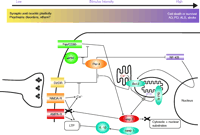
Interactions of apoptosis pathways with calcium signaling and synaptic transmission. Par-4 can transduce classical apoptosis pathways like Fas/CD95-mediated cell death by interaction with atypical PKC (ζ) or by inhibiting NF-κB–mediated expression of Bcl-2. At the same time, Par-4 interacts with the D2 dopamine receptor at synapses to inhibit downstream dopamine signaling. This latter interaction is antagonized by calcium-activated calmodulin. Bcl-2, apart from its most typical action, inhibiting the formation of the mitochondrial permeability transition pore, also enhances calcium leakage from the endoplasmic reticulum. Activated caspase-3 can cleave GluR1, an α-amino-3-hydroxy-5-methylisoxazole-4-propionate (AMPA) receptor subunit, thereby modulating intracellular calcium levels. → indicates stimulation and ⊣ indicates inhibition. D2DR, dopamine 2 dopamine receptor; NMDA-R, NMDA receptor; AMPA-R, AMPA receptor, atPKC, atypical PKC (ζ), LTP, long term potentiation; IL-1, interleukin-1β; casp 1, caspase 1; casp 3, caspase 3; PTP, mitochondrial permeability transition pore; NF-κB, Nuclear factor-kappaB; AD, Alzheimer Disease; PD, Parkinson Disease; ALS, amyotrophic lateral sclerosis.
- © American Society for Pharmacology and Experimental Theraputics 2005
References

Mark P. Mattson, PhD, (right) received his graduate training from the University of Iowa and postdoctoral training in developmental neurobiology at Colorado State University. He then joined the faculty at the Sanders-Brown Center on Aging at the University of Kentucky where he studied neuronal plasticity and the pathogenesis of neurodegenerative disorders. In 2000, Dr. Mattson became Chief of the Laboratory of Neurosciences at the National Institute on Aging Intramural Research Program in Baltimore. He is also a Professor in the Department of Neuroscience at the Johns Hopkins University. Dr. Mattson’s laboratory investigates the molecular and cellular mechanisms responsible for neuronal dysfunction and death in age-related neurological disorders and is working to establish novel preventative and therapeutic interventions for these disorders. Please address correspondence to MPM. E-mail mattsonm{at}grc.nia.nih.gov; fax(410) 558-8465. Marc Gleichmann, MD, PhD, (left) completed medical school at the University of Duesseldorf, Germany, where he also received his graduate training in the lab of Hans-Werner Mueller, working on sciatic nerve regeneration. He received clinical training in neurology at the University of Tuebingen where he worked in the lab of Joerg B. Schulz on neurodegeneration and apoptosis. Dr. Gleichmann joined the laboratory of Dr. Mattson in 2004 where he is a Postdoctoral Fellow. E-mail gleichmannma{at}grc.nia.nih.gov

