Coregulators in Nuclear Estrogen Receptor Action
From Concept to Therapeutic Targeting
Abstract
Estrogens are key regulators of growth, differentiation, and the physiological functions of a wide range of target tissues, including the male and female reproductive tracts, breast, and skeletal, nervous, cardiovascular, digestive and immune systems. The majority of these biological activities of estrogens are mediated through two genetically distinct receptors, ERα and ERβ, which function as hormone-inducible transcription factors. Over the past decade, it has become increasingly clear that the recruitment of coregulatory proteins to ERs is required for ER-mediated transcriptional and biological activities. These “coactivator” complexes enable the ERs to respond appropriately: 1) to hormones or pharmacological ligands, 2) interpret extra- and intra-cellular signals, 3) catalyze the process of chromatin condensation and 4) to communicate with the general transcription apparatus at target gene promoters. In addition to activating proteins, the existence of corepressors, proteins that function as negative regulators of ER activity in either physiological or pharmacological contexts, provides an additional level of complexity in ER action. This review also describes current efforts aimed at developing pharmaceutical agents that target ER-cofactor interactions as therapeutics for estrogen-associated pathologies.

Molecular Mechanisms Of Er And Associated Coactivators
Principles of ER-action in the Nucleus
The ERs (ERα and ERβ) are Class I members of the nuclear receptor (NR) superfamily of ligand-inducible transcription factors (1). According to the classical model of ER action, in the absence of hormone, the receptor is sequestered in a multiprotein inhibitory complex in either the cytoplasm or nuclei of target cells. The binding of ligand induces an activating conformational change within the ER, promoting dimerization and high-affinity binding to specific estrogen-response elements (EREs) located within the regulatory regions of target genes. From these sequences the ER communicates with the general transcription apparatus to positively or negatively regulate gene transcription.
ERα and ERβ possess the hallmark modular structure characteristic of other NRs (1) (Figure 1⇓). Because ERα and ERβ display a high degree of sequence similarity in the central DNA- and C-terminal ligand-binding domains (DBD, 97% and LBD, 60%, respectively) (Figure 1⇓), it is not surprising that these receptors interact with identical response elements and exhibit similar binding affinity profiles for an array of endogenous, synthetic, and naturally occurring estrogens (2, 3). Thus, it was initially speculated that ERβ might be a functionally redundant form of ERα, although its tissue distribution suggested otherwise. We now know that ERα is widely expressed and is the predominant ER subtype in the breast, uterus, and bone. On the other hand, ERβ is expressed primarily in the ovary, prostate, testis, lung, thymus, spleen, and in localized areas of the brain (4). Not surprisingly, ERα mediates the physiological actions of estrogens in mammary gland development, maintenance of bone mineral density, and mating behavior, as well as in glucose metabolism, the cardiovascular system, and the hypothalamic-pituitary axis. On the other hand, ERβ is involved in regulating ovulation, some aspects of mating behavior, and immune responses (5). Intrinsic mechanistic differences may also distinguish the two receptors, as it was shown that ERβ could function as an inhibitor of ERα, reducing the potency of estrogens acting through ERα (6). The unique functional activities of the two ERs have been confirmed by using animal models that are deficient in one or both receptors and by recently developed receptor-selective agonists and antagonists (5, 7).
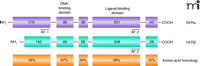
Functional domains of the human estrogen receptors. ERα and ERβ share a highly conserved central DNA-binding domain and moderately conserved C-terminal ligand-binding domain. The ligand-dependent transcriptional activities of the ERs are mediated through a C-terminal activation function (AF-2). ERα contains a constitutive AF-1 in the N terminus; no apparent AF-1 domain is present in the human ERβ (6). Numbers in boxes indicate the number of residues in each protein region.
Two acidic activation domains mediate the ligand-dependent transcriptional activity of ERα: a constitutive activation function-1 (AF-1) in the N terminus, and a hormone-dependent AF-2 located in the ligand-binding domain (Figure 1⇑). The AFs function in a synergistic manner in most circumstances but may also function independently in certain cell and promoter contexts (8). The activation domains in ERβ are less well characterized. Sequence similarity and functional studies indicate that ERβ also contains an AF-2 (located in the C-terminal portion of its LBD); however, reports that this region functions as the predominant regulator of transcription by ERβ imply the AF-2 domains of the two ER subtypes play distinct roles. Furthermore, given the high degree of sequence divergence and the lack of an obvious AF-1 domain in the human ERβ subtype, it is not surprising that the two receptors have been found to be functionally distinct (6). Thus, mechanistic differences in the activation domains of the two ER subtypes may underlie their distinct biological roles in estrogen signaling.
In addition to the classical (ligand- and ERE-mediated) pathway, there are at least two other mechanisms by which the ERs can regulate the transcriptional activities of their respective target genes. First, the observation that polypeptide growth factors such as epidermal growth factor (EGF) and insulin-like growth factor 1 (IGF-1) and the intracellular effector cAMP activate the ER and increase expression of ER target genes led to recognition of the roles of ligand-independent receptor activities in estrogen biology (9). Furthermore, studies reporting estradiol-ER induction of genes containing no apparent ERE-like sequence led to the discovery that ligand-activated ER can interact in an indirect manner with the regulatory regions of target genes. Specifically, ERα-mediated expression of the collagenase and IGF-1 genes is mediated through the interaction of the receptor with Fos and Jun at AP-1 binding sites, whereas several genes containing GC-rich promoter sequences are activated through ERα-Sp1 complexes (10). The role of these “non-classical” modes of nuclear action will be discussed in more detail below in the context of coregulators and ER pharmacology.
Evidence for the Existence of ER Modulatory Factors
The observation that the transcriptional activities of ER ligands are manifested in a tissue-selective manner suggests that the receptor does not function in isolation, but rather, requires specific cellular factors for maximal responses. Evidence for an interaction between activated NRs and proteins other than the general transcription apparatus arose from studies that probed the phenomenon of transcriptional interference (i.e., squelching). This term refers to the mechanism by which the activity of an activated receptor at its target promoter can be suppressed by overexpression of a distinct, though related transcription factor. Specifically, the yeast Gal4 transcription factor could negatively regulate expression of specific genes in a manner that did not require the Gal4 target promoter or DNA binding (11). Similarly, ER was shown to inhibit progesterone receptor (PR) and glucocorticoid receptor (GR) activation on promoters lacking EREs (12). Collectively these observations suggested that the sequestration of a limiting cellular pool of factors required by both transactivators was responsible for reducing their transcriptional responses. These studies led to subsequent identification of a whole host of cellular proteins that interacted with the liganded LBD of ER and several NRs, and with other transcription factors.
Identification of ER AF-2 Coactivators
The first ER-interacting proteins were identified by Halachmi et al., who used the purified, liganded ERα LBD to isolate two proteins of approximately 140 and 160 kilodaltons, termed ER-associated proteins 140 and 160 (ERAP140, ERAP160) (13). These particular receptor-cofactor associations were agonist-dependent and required an intact AF-2 domain. Although ERAP140 and ERAP160 interacted with several NRs, they did not associate with other transcription factors, indicating some degree of specificity. Subsequent studies identified additional proteins that bound the LBD of ER and other NRs, including transcriptional intermediary factor 1 (TIF-1), receptor interacting proteins 140 and 160 (RIP140, RIP160), human receptor potentiating factor 1 (hRPF1), thyroid hormone receptor interacting protein 1 (TRIP1/SUG1), and thyroid hormone receptor associated proteins (TRAPs/DRIPs) (14). It was not until the cloning and characterization of the coactivator SRC-1 (steroid receptor coactivator-1) was accomplished, however, that a specific role for this class of proteins in ER action was defined. The SRC-1 protein was initially identified in a yeast two-hybrid screen, using the PR LBD as bait, and was subsequently shown to interact with ERα and potentiate its activity in an agonist-dependent manner (15). The observation that SRC-1 could reverse squelching of PR transcriptional activity by ER suggested that it was a limiting factor required by both receptors. SRC-1 interacts with several other NRs and enhances their transcriptional activities in the presence of activating ligands. The cloning of SRC-1 led to the discovery of a whole family of structurally and functionally similar coactivators (termed the p160s). These include the murine glucocorticoid receptor-interacting protein-1 (GRIP1), transcriptional intermediary factor 2 (TIF-2; human homolog of GRIP1), and amplified in breast and ovarian cancer-1 (AIB-1) (Table 1⇓). Many of these cofactors contain intrinsic histone acetylase activity (HAT), which is known to facilitate chromatin remodeling at target promoters (16, 17).
Coactivators in ER Physiology and Pharmacology
Receptor-Coactivator Interactions Are Mediated Through ER Helix 12 and the LXXLL Motifs of Coactivators
A series of structure-function studies have led to the elucidation of the mechanisms governing coactivator recruitment by the AF-2 regions of NRs. Specifically, the LBD of ER and other NRs contain twelve α-helices, designated H1–H12. In the presence of agonist, the AF-2 pocket is formed by the folding of H12 against helices 3, 5/6, and 11 (18). That the position of H12 is different in agonist-bound ER, antagonist-bound ER, and apo-ER suggests that H12 is an important regulator of coactivator recruitment. Crystallization of the ERα LBD with a peptide from the GRIP1 NR interaction region revealed that coactivators bind to a hydrophobic groove on the surface of the LBD that is exposed upon the repositioning of H12 in the presence of agonists. In the antagonist-occupied receptor, however, H12 occupies the coactivator binding surface, mimicking the coactivator interaction and thus blocking cofactor recruitment (19). In parallel with these studies, the NR interacting regions of coactivators have been characterized. Heery et al. identified a conserved motif within SRC family members called the nuclear receptor (NR) box (LXXLL; L = leucine, X = any amino acid), which is necessary and sufficient for coactivator binding to activated receptors (20). These structural motifs form amphipathic α-helices in which the leucines create a hydrophobic surface that fits into the hydrophobic groove of the receptor AF-2 domain. Three LXXLL motifs are conserved within SRC family members, and several additional NR boxes are found in other coactivators, including TRAP220, CREB-binding protein (CBP), p300, and the activating signal cointegrators (ASC-1 and ASC-2) (Table 1⇑). The apparent functional redundancy in coactivators and the finding that they share a conserved NR interaction surface raised the question as to how specificity is achieved. Ensuing studies revealed that the sequences flanking the LXXLL motifs are key determinants of the binding affinity and specificity of coactivators for their NR partners (21, 22). Additionally, multiple NR boxes within a single coactivator contribute to the selectivity of interaction; whereas a single NR box in SRC-1 is sufficient for ERα activity, different combinations of two motifs are required for PR, thyroid hormone receptor (TR), and retinoic acid receptor (RAR) activity (22). Collectively, these studies provide insight into the mechanisms regulating the recruitment of AF-2 coactivators by ER and other NRs.
Coactivators Grouped By Binding Characteristics
ER AF-1 Coactivators
In addition to the coactivators that enhance receptor activity by interacting with the LBD, AF-1-interacting coactivators have also been described (Table 1⇑). Specifically, steroid receptor RNA activator (SRA) is an RNA transcript that enhances the AF-1 activity of ER and other steroid receptors (23). Endoh et al. have characterized a p68 RNA helicase that potentiates the activity of ERα AF-1 in both the estrogen- and antiestrogen (i.e., estrogen antagonist)-liganded receptor (24). Interestingly, this cofactor enhances the transcriptional activity of ERα, but not ERβ, perhaps reflecting the absence of a functional AF-1 in the human ERβ.
Secondary ER Coactivators
In addition to coactivators that bind the ERs in a direct manner, a series of factors have been described that enhance ER activity through their ability to interact with the p160 coactivators, thus affecting ERs indirectly (Table 1⇑). For instance, the p160s contain not only LXXLL motifs that allow for ER binding but also contain C-terminal activation domains (AD1 and AD2) and N-terminal basic-helix-loop-helix/PAS (bHLH/PAS) domains, which associate with factors involved in chromatin remodeling. Specifically, AD1 recruits the histone acetyltransferases CBP and p300, and AD2 interacts with protein arginine methyltranferases (PRMTs) such as coactivator-associated arginine methyltransferase 1 (CARM1) and PRMT1. The existence of these secondary complexes allows for amplification of ER responses: SRC-1 together with CBP, and GRIP1 together with PRMTs were shown to function in a synergistic manner to potentiate the transcriptional activity of ER (25). Furthermore, a recently described ER cofactor, coiled-coil coactivator (CoCoA), which binds the bHLH/PAS domain of p160s, enhances ER target gene expression by associating with p160s (26).
Dual Functional Coactivators
A series of cofactors has recently been characterized that displays activities in addition to those related to direct enhancement of ER transcription. E6-associated protein (E6-AP) and receptor potentiating factor-1 (RPF-1) are ubiquitin protein ligases that potentiate the ligand-dependent transcriptional activity of ER (Table 1⇑). Although the ubiquitin ligase activities are dispensable for their coactivator function, the ability of E6-AP and RPF-1 to ubiquitinate proteins prior to degradation by the 26S proteasome (27) may enhance transcription of ER target genes by catalyzing the dynamic exchange of ER, cofactors, and other transcriptional components (discussed below).
Peroxisome proliferator-activated receptor γ coactivator-1 (PGC-1), which was originally identified as a transcriptional coactivator of the peroxisome proliferator-activated receptors (PPARs), displays physical and functional interactions with several NR family members including the ERs. Specifically, PGC-1 binds the hinge region in ER, located between the central DNA-binding and C-terminal ligand-binding domains. Cell-based assays demonstrated that PGC-1 potentiates the transcriptional activities of ER through AF-1 and/or AF-2 in a cell and promoter context-dependent manner (28). PGC-1 also contains an RNA recognition motif that is required for this cofactor to stimulate the expression of intron-containing target genes and to influence the choice of alternative splicing sites (29).
O’Malley and colleagues have also provided compelling evidence that shows transcription and pre-mRNA processing are functionally coupled: steroid hormones may affect alternative splicing of pre-mRNAs synthesized from steroid-responsive promoters in a receptor-dependent manner. Three recently identified coactivators, Coactivator of activating protein-1 and estrogen receptors (CAPER-alpha, CAPER-beta) and coactivator activator (CoAA), integrate the potentiation of steroid receptor transcription and alternative splicing of pre-mRNAs through distinct and separable domains in the proteins (Table 1⇑) (29, 30). The apparent promoter-specific activities of the splicing and transcription functions may provide a mechanism for achieving additional selectivity in estrogen-mediated gene expression by enhancing ER activity and processing ER target gene pre-mRNAs in a gene-discriminatory manner.
Identifying Corepressors of ER
Dampening Estrogen-ER Action
The fact that all known natural ligands of ERs are agonists suggests that the cellular role of NRs is to elevate transcription from target gene promoters; hence, the existence of coactivator proteins that amplify these responses is reasonable. Physiologically, however, there are fluctuating levels of circulating estrogens, and in tissues such as ovary, chronically high levels of estrogen could provide for sustained ER activation and overstimulation of ER biological pathways. Thus, as coactivators enhance ER activity, corepressor proteins have been identified that dampen the agonist effects of estrogens. A recently described corepressor of ERα and ERβ, termed repressor of estrogen receptor activity (REA), decreases ER activity by interfering with SRC-1 access to the receptor (Table 2⇓) (31). RIP140, an LXXLL-motif containing protein, associates with the estrogen-bound ERs through the AF-2 domain, occluding access of AF-2 coactivators. In addition, RIP140 can decrease basal ER target gene expression by associating with histone deacetylase (HDAC) complexes, which repress transcription by catalyzing the condensation of chromatin (29). Thus, the existence of corepressors that moderate the agonist activities of estrogens provides an additional mechanism for fine-tuning the expression of ER target genes and attentuating the physiological output in situations where there are chronically elevated levels of hormone.
Corepressors in ER Physiology and Pharmacology
Pharmacological Corepressors of ER
The first identified and perhaps most-studied corepressors in NR biology are the nuclear receptor corepressor (NCoR) and silencing mediator for retinoid and thyroid hormone receptors (SMRT) (Table 2⇑). These proteins were originally identified as transrepressors of several of the nonsteroid nuclear hormone receptors such as thyroid hormone receptor (TR) and retinoid activated receptor (RAR), which occupy their target regulatory DNA sequences and repress transcription in the absence of hormone (rather than existing in inactive complexes, as the ER and PR do with heat shock proteins). NCoR and SMRT are functionally similar as they interact with unliganded receptors, enhance repression, dissociate upon agonist binding, and contain intrinsic silencing domains (32, 33). The interaction of these corepressors with NR LBDs is mediated by two NR-interacting domains (CoRNR boxes) found within both NCoR and SMRT, which contain sequences that are similar to those found in the NR boxes (LXXLL motifs) of coactivators (34).
Like coactivators, NCoR and SMRT interact with secondary cofactors that enable them to manifest their repressive effects on NRs. The repressor domains of NCoR and SMRT bind mSin3, a protein that associates with HDACs (35). Tissue-specific repression of NRs is also mediated by a secondary cofactor, mSiah2, which targets NCoR for proteasomal degradation. The amount of mSiah2 found in cells varies according to the cell type, and its expression is inversely correlated with cellular NCoR content and degree of NR transcriptional repression (36).
Whereas NCoR and SMRT bind and repress the transcriptional activities of many NRs and unrelated transcription factors, the physical and functional interaction of these corepressors with the ERs is limited to pharmacological contexts. Thus, their specific role in ER action is discussed later in this review (see Cofactors and ER Pharmacology).
Corepressors exist that modulate ER pharmacological activities through regions other than the receptor LBD. Norris et al. identified an RNA binding protein called “repressor of tamoxifen transcriptional activity” (RTA) that interacts with the N terminus (AF-1) of ERα. It is interesting to note that RTA inhibits the partial agonist activity of tamoxifen (a synthetic ER ligand) through the ERα while displaying minimal effects on estrogen-mediated transcription (Table 2⇑) (37). This observation suggests that synthetic ER ligands may manifest their biological effects in part by enabling the receptor to interact with proteins other than those utilized in a physiological context.
In summary, it is now clear that the transcriptional activities of the ERs are regulated by a vast array of cellular proteins, including those discussed above (Figure 2⇓) and others (14, 16, 27, 29). This complex network of coactivators and corepressors provides for balanced, sensitive control of ER target gene expression.
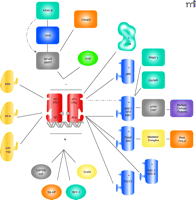
A network of cofactors in nuclear ER action. ERs utilize a complex network of coactivators and corepressors that provide for balanced, sensitive control of ER-mediated target gene expression [Figure modified from (16)].
Regulation of ER-Cofactor Complexes
Cell Signaling
In addition to ligand-mediated receptor pathways, ER can be activated by extracellular signals and enhance target gene expression in the absence of hormone. The observation that the activation domains of the ERs are also required for ligand-independent receptor function (i.e., AF-1 for growth factor activation and AF-2 for cAMP activation) suggested a role for classical ER coactivators in these pathways (9). Indeed, mitogen-activated protein (MAP) kinase-induced activation of the ERα AF-1 by phosphorylation of the Ser118 residue is concomitant with recruitment of the AF-1 coactivator p68 RNA helicase following treatment with EGF or IGF-1 (24). Furthermore, growth factor stimulation enhances phosphorylation of the murine ERβ AF-1 and promotes SRC-1 binding and transcriptional activation (38). It is also apparent that cell signaling pathways can facilitate coactivator-corepressor exchange, as cAMP stimulation of mammalian cells promotes dissociation of NCoR and SMRT from antagonist-bound PR, allowing for coactivator access (39).
Not all of the ligand-independent activities of ERs affected by extracellular pathways can be attributed to direct kinase phosphorylation of the receptors. Given that most cofactors exist in similar amounts in all tissues, it is reasonable to anticipate that there may exist mechanisms to turn on and off their activities to provide more sensitive control of transcription. Accordingly, coactivators were recently found to serve as points of convergence between ER and growth factor pathways: SRC-1, GRIP1, and AIB1 are phosphorylated by MAPK, an event that enhances their activities (29). More elaborate control of coregulator function was demonstrated with the identification of six phosphorylation sites in AIB1––all of which are required for coactivation of ER––yet different combinations of phosphorylation sites on AIB1 are required for mediating the activation of NF-κB compared to phosphorylation patterns on AIB1 required for oncogenic transformation of MEFs (40). Current evidence indicates that phosphorylation of cofactors may enhance their activities; however, MEK-1-mediated phosphorylation of SMRT effectively causes a loss of repression by promoting disassociation of SMRT-TR complexes (41). Thus, extracellular signaling pathways may enhance NR action by coordinately enhancing the recruitment and activity of coactivators while decreasing the association and functionality of corepressors.
Posttranslational Modifications
In addition to phosphorylation, other types of posttranslational modifications have been identified that regulate coregulator function. Acetylation and ubiquitination can decrease the half-life of cofactors in ER transcriptional complexes by catalyzing their dissociation or degradation. Other modifications may enhance transcriptional functions; methylation of CBP by CARM1 is important for strong CBP coactivation of ER and GRIP1 complexes, and sumoylation (addition of a small ubiquitin-like modifier) of SRC-1 or GRIP1 enhances coactivation functions by retaining these cofactors in the nucleus (27, 29). Overall, posttranslational modification of cofactors appears to provide a mechanism to integrate extracellular signaling pathways, regulate assembly and dissociation of coregulators, and enhance or decrease the transcriptional efficacy of ER-cofactor complexes.
Regulation by DNA and Promoter Context
Although a regulatory role for ligand and cofactor association in ER transactivation has been well-defined, there is emerging evidence that specific DNA response elements function as allosteric ligands for NRs, altering their structure and function (42). Using a series of naturally occurring EREs, it was shown that the nature of the response element alters the structure of the ER coactivator-binding pocket in a manner that influences coactivator recruitment and transcriptional activation (43). In addition to promoter-specific cofactor requirements, ER promoter context may in fact effect functional conversion of corepressors to coactivators and vice versa (27).
The role of promoter-context in regulating ER-cofactor association was also shown in a comparison of ERE-mediated vs nonclassical modes of receptor action. A biochemical assay on chromatin templates recently demonstrated that there are both common and dissimilar ER coactivator preferences in ERE- vs. AP-1-mediated ER signaling. Specifically, SRC-p300/CBP interactions were required for both pathways, whereas transactivation through the ER-ERE pathway was substantially more dependent on p300/CBP acetyl-transferase activity and TRAP220 recruitment by ER (44). Thus, the type of enhancer utilized may affect the assembly of ER-coregulatory complexes and transcriptional activation, providing a mechanism for the gene-specific activities of the ERs.
Cyclical and Sequential Control of Complex Activation on ER Target Promoters
An additional level of complexity in ER-cofactor assembly on target gene promoters was recognized with the introduction of chromatin immunoprecipitation (ChIP) assay technology. Initial studies demonstrated that there is sequential and cyclical recruitment of coactivators to the estrogen-responsive pS2 gene promoter, with ERα being the first to be recruited, followed by AIB1, p300 and TRAP220, and then RNA polymerase II (45). More detailed analysis of this promoter revealed subsequent assembly of ERα and coregulator-complexes containing chromatin remodeling, methylation, acetylation, and ubiquitination activities (46). Displacement of this preinititiation complex occurs by proteasomal degradation of the ER, allowing for promoter clearance and reinitiation of subsequent rounds of transcription. These studies demonstrated that for efficient ER target gene transcription there is a clear requirement for: 1) coactivators such as E6-AP and p300 (which ubiquitinate ER to direct it for degradation), 2) cofactors with diverse enzymatic activities, and 3) precise and ordered association and dissociation of cofactor complexes with ER-bound promoters (27, 29, 45, 46).
Cofactors and ER Biology
Role of Cofactors in ER-Mediated Physiological Processes
Based on the essential roles of cofactors in ER activity, it was initially hypothesized that the relative expression levels of ER coregulators in different tissues would significantly impact estrogen biology. The expression patterns of most cofactors examined to date, however, is broad (with some exceptions). The most well studied coregulators, the p160s, NCoR, and SMRT, have been detected in most cell and tissue types. On the other hand, PGC-1 shows perhaps the most tissue-restricted expression, with PGC-1 mRNA detected in heart, muscle, kidney, and liver, yet absent in other ER target tissues such as brain, colon, lung, intestine, and thymus (29).
The development of mice harboring genetic knockouts (KOs) for individual cofactor proteins, including the p160s (SRC-1, SRC-2/GRIP1, and SRC-3/AIB1), E6-AP, RAP250, TRAP220, CBP, p300, NCoR, RIP140, and REA has been informative as to the unique and overlapping roles of these cofactors in estrogen biology. Of the three different p160 single KO strains, only SRC-1-null mice have a phenotype of generalized resistance to steroid hormones. As a consequence, estrogen-dependent proliferation of mammary ductal branches is seriously compromised in these animals, indicating that the ability of this cofactor to modulate ER function may effectively enhance physiological sensitivity to hormone (47). SRC-2 (GRIP1)-null mice are distinguished from the other p160 KOs in that they display impaired fertility and gonadal function (48). On the other hand, only SRC-3 (AIB1) KO animals exhibit deficient overall growth and reduced mammary gland development (49); thus, even structurally related coactivators are not functionally equivalent. Despite these observations, it is clear that in some instances cofactors can compensate for one another, as was demonstrated in SRC-1/SRC-2 compound mutant mice, where SRC-1 can partially compensate for the effects of a loss of SRC-2 on mouse survival and growth (50).
Studying the in vivo biology of many other NR cofactors has proved more challenging, as homozygous deletion of CBP, p300, TRAP220, ASC-2, NCoR, or REA is lethal (29). This perhaps reflects their ability to regulate a wider range of NR and transcription factor functions. Interestingly, however, even in REA heterozygote animals that express half of the amount of REA found in wild-type (wt) animals, a clear role for this cofactor in estrogen uterine biology was defined. When compared to wt animals, REA heterozygous mice show remarkably enhanced uterine cell proliferation and increased overall uterine growth response to estrogen. Microarray studies revealed that these phenotypic changes are reflected at the genomic level with an estrogen-dependent increase in ER target gene expression and loss of decreased expression of genes normally repressed by estrogen (51). These observations that REA restrains estrogen action by moderating stimulation and enhancing ER repression of target genes indicate that REA is involved in regulating tissue sensitivity to estrogens.
Another corepressor with a potentially important role in estrogen biology is RIP140. Mice with homozygous deletion of RIP140 display impaired fertility attributed to compromised ovarian function (52), suggesting a role for this corepressor in ERα and/or ERβ action. Interestingly, RIP140 KO animals are lean and exhibit enhanced energy expenditure in contrast to ERα KO mice, which display increased body weight and adiposity compared to wt littermates (53). These observations suggest a role for ERα-RIP140 signaling in lipid homeostasis and metabolism, providing a potential mechanism by which energy storage and expenditure are balanced. This hypothesis, however, will require much closer examination, as RIP140 interacts with and represses the activities of many NRs, some of which (such as the PPARs and ERRs) are also known to regulate lipid homeostasis and metabolism.
Cofactors in ER Pathophysiology
In addition to the ability of cofactors to regulate ER physiology, these proteins also play an important role in estrogen-associated pathologies. The observed phenotype of SRC-1 KO mice, which display substantially compromised hormone-dependent proliferation of mammary ductal branches, suggested that alterations of cofactor levels might occur in hormone-dependent cancers. Commensurate with this observation, data implicating coactivator overexpression in human breast cancer pathogenesis, whether as a cause or effect, have been steadily accumulating. Elevated amounts of SRC-2 and CBP have been observed in intraductal carcinomas as compared to those amounts found in normal mammary tissue, and overexpression of TRAP220 and E6-AP has been observed in breast tumors (54–56). Perhaps most notably, the p160 family member AIB-1/SRC-3 is overexpressed in 60% of primary breast tumors, and amplification of the gene seems to be a consistent feature of up to 10% of breast tumors (57). The pathological consequences of aberrant AIB1 levels were recently demonstrated in transgenic mice in which over-expression of AIB1 in the mammary gland resulted in development of malignant mammary tumors (58). In fact, elevated expression of AIB1 in breast and ovarian cancers is coupled with hyperactivated growth factor kinase pathways (i.e., HER2/neu), which target AIB1 and other cofactors, enhancing their activities on the ER. Thus, it has been proposed that coactivator overexpression and kinase superactivation are part of an integrated mechanism in promoting carcinogenesis (59).
Although controversial, some studies also suggest that alteration in the expression of coactivators and corepressors may contribute to antiestrogen-resistance in breast cancer. In a mouse model for human breast cancer, NCoR amounts were decreased in antiestrogen-resistant tumors as compared to NCoR amounts found in tumors that did respond to the drug (60). Further evidence was found in human invasive breast tumors, where overexpression of AIB1 and HER2 together was associated with tamoxifen resistance and lower survival rate (59).
Estrogen-dependent cancers represent the most widely studied disease models of cofactors in ER pathology; however, the recognition that ER functions in a wide variety of target tissues suggests a more global role for cofactors in disease. One can speculate that alterations in cofactor levels could be associated with heart disease, osteoporosis, impaired fertility, neurological disorders, and compromises in immune system function; this hypothesis awaits formal evaluation. Regardless, it is anticipated that the implementation of new genetic screening technologies will allow for identification of genetic allelic variations in NRs and cofactors that may predispose people to diabetes, obesity, heart disease, cancer, and other pathologies.
Cofactor-Receptor Pairing
As put forth above, one the greatest obstacles in identifying specific roles of different coregulators in ER physiology and pathology is the ability of most of these factors to regulate the activities of several NR family members. To clearly define the biology of cofactors in ER action will require a reductionist approach to enable isolation and definition of the activities of specific ER-cofactor pairs in mammalian cells and in vivo. Recognizing an unmet need for such an approach, our laboratory has recently developed a new technology that will enable forthcoming functional assessment of specific ER-coregulator pairs in vitro and in vivo.
Cofactors and ER Pharmacology
Pharmacological Modulation of ER Activity by Antiestrogens and SERMs
The multitude of tissue-selective biological effects mediated by the ERs has necessitated therapeutic development of synthetic agents with mixed agonist-antagonist effects on the ERs. The identification of compounds that retain the beneficial effects of estrogen in the bone, brain, cardiovascular, and immune systems, yet lack the mitogenic and perhaps carcinogenic activities in the breast and uterus, continues to be a major challenge to the pharmaceutical industry. The existence of such tissue-selective ER modulators was not initially predicted, as according to classical receptor theory, agonists function as molecular switches, converting ER from an inactive to an active form whereas antiestrogens competitively inhibit agonist binding and lock the receptor in a latent state. Clearly, however, this model was oversimplified, as it does not account for tissue-selective agonist and antagonist activities of several known antiestrogens (61). The first evidence that the activities of synthetic ER ligands could be dependent on the target tissue came from clinical studies in which patients that were administered tamoxifen as adjuvant therapy for estrogen-dependent breast tumors exhibited estrogen-like effects on bone density (62). Since this initial observation, there has been a wealth of experimental evidence demonstrating that agonists and antagonists induce distinct structural alterations in the ERs. Thus, both physiological and pharmacological ER ligands play active, yet distinct roles in modulating receptor conformation and function (2, 16, 29, 63).
Based on structural and functional data, three classes of anti-estrogens were initially defined (64). Type I antiestrogens, such as ICI 182,780, function as pure ER antagonists and oppose estrogen activity in all tissues. Type II and type III antiestrogens, represented by raloxifene and tamoxifen, respectively, have been defined as selective estrogen receptor modulators (SERMs), which differ from pure antagonists in their capacity to display tissue-selective agonist-antagonist activities (61, 65, 66). Many of these agents are in clinical use; the antagonist activity of tamoxifen in the breast makes it a first line therapy for pre- and post-menopausal women with advanced breast cancer. Unfortunately, however, the agonist activity of tamoxifen is manifest in the uterus and is likely to be involved in the process by which tumors alter their physiology to become tamoxifen-resistant and recognize the drug as an agonist for growth (61, 65, 66). The finding that tamoxifen-resistant breast cancer xenographs are not cross-resistant to GW5638 (a tamoxifen-derived compound), however, suggests the potential to develop new tissue-targeted SERMs that retain the beneficial effects of estrogen in some tissues while functioning as antagonists in the breast and uterus (67).
Corepressors Mediate the Antagonist Activities of Antiestrogens
The discovery of ER antagonists with unique biological activities, and which were capable of inducing distinct conformational changes in the ERs, prompted an evaluation of the role of cofactors in antiestrogen pharmacology. As stated above, estrogen binding to ER induces a conformational change in the receptor that permits the formation of a hydrophobic pocket in the AF-2 domain that facilitates coactivator recruitment through their LXXLL motifs (18, 20). Conversely, antiestrogens function as AF-2 antagonists, in that the structural changes in ER they induce do not permit the binding of the LXXLL-interaction motif (19). It is now clear, however, that the antagonist activities of antiestrogens are not solely attributed to the inability to promote assembly of ER-coactivator complexes. The observation that antiestrogen-ER complexes bind DNA suggested that they might be able to mediate active repression by recruiting corepressors to target promoters. Likewise, roles for the corepressors NCoR and SMRT in mediating the antagonist activities of both pure antiestrogens and SERMs has been recently demonstrated (29). Detailed characterization of ER-corepressor interactions using CoRNR box peptides, however, suggested that corepressors other than NCoR and SMRT may also involved in ER pharmacology (68). A possible candidate is RTA, which suppresses the partial agonist activity of tamoxifen by interacting with the N terminus of ERα (37).
Coactivators and SERM Agonist Activity
Despite a tremendous amount of research effort, it is still not entirely clear how SERMs manifest their partial agonist activities through the ER. The ability of these compounds to antagonize AF-2 coactivator association suggested that other receptor domains and cofactors could be responsible for SERM activity. Indeed, tamoxifen partial activity requires the N-terminal AF-1 of ERα and is potentiated by the AF-1 cofactor p68 RNA helicase (24). AF-2 coactivators may also be involved in SERM action in some circumstances, because overexpression of SRC-1 enhances the agonist activity of tamoxifen in HepG2 (liver) cells where tamoxifen displays substantial partial agonist activity (69). Similar effects of p160s were not observed in all cell types, indicating that cell context can influence the ability of SERM-bound ER to utilize cofactors.
The discovery that SERMs such as raloxifene and GW5638 function as estrogen agonists in bone and the cardiovascular system but do not appear to function as ER AF-1 or AF-2 agonists on ERE-containing promoters suggested that not all of the biological activities of SERMs are mediated through classical ER pathways (2, 61). Evidence has emerged that SERMs function as effective ER agonists at AP-1-responsive promoters, and these activities have distinct cofactor requirements as compared to SERM-bound ER complexes at ERE-containing promoters (44, 70). Extracellular signaling pathways mediated by growth factors or cAMP treatment can also regulate the agonist activities of SERMs by facilitating either AF-1 or AF-2 coactivator recruitment to the ERs (29). Overall, it is now appreciated that coregulators function as an integral part in SERM activity by their ability: 1) to distinguish changes in ER conformation, sense the DNA and tissue context, and respond to cell signaling events, and 2) to integrate all of these variables to mediate ER agonist or antagonist activity.
Tissue-Selective Agonist-Antagonist Activities of SERMs Mediated by Different Coactivator-Corepressor Recruitment
Together, the discovery of SERMs, and identification of ER coregulators with distinct activities, suggested that the agonist-antagonist activities of antiestrogens could be mediated through differential coactivator-corepressor recruitment in certain cell and promoter contexts. In fact, coexpression of ERα with coactivator or corepressor alters tamoxifen agonist-antagonist activities (69). The observation that antagonist-liganded PR functions as a transactivator when coexpressed with tamoxifen-bound ER or unliganded TR (71) suggested that corepressors may function as a switch, converting a potentially active receptor into an inert or repressive state. It has also been hypothesized that the relative expression of coactivators and corepressors in ER target cells may be a primary determinant of the agonist-antagonist activities of SERMs (2, 29, 61). Thus, one could envision that tamoxifen resistance in ER-positive tumors could occur as a result of alterations in cellular levels of cofactors, a hypothesis currently under investigation (see above in Cofactors and ER Biology).
Targeting ER-Cofactor Interactions to Develop New Therapeutics
Peptide Conformational Probes Facilitate ER Drug Discovery
Cell-based reporter assays have been used extensively for NR lead compound discovery, in which ligands are evaluated according to their ability to activate transcription from a hormone response element fused to a luciferase reporter gene (72). Given the complexity of ER action [i.e., dependence on ER subtype, tissue and promoter context and cell signaling events], however, it is unlikely that a simple reporter gene assay would be sufficient to discriminate among structurally similar, yet mechanistically distinct agents. For example, the structurally related SERMs tamoxifen and GW5638 assayed in this system would be classified together as ER antagonists with similar efficacies (because of their comparable receptor affinities) despite the fact that the two agents display unique activities: GW5638 lacks agonist activity in the uterus and can counteract the agonist activity of tamoxifen in hormone-resistant breast tumors (61, 67).
As put forth above, the structures of the estrogen-ER and SERM-ER complexes are distinct. These conformational states mediate the cellular responses to agonists and antagonists by regulating the interaction of the ERs with coactivators and corepressors. Given these findings, it was proposed that different ligands expose different receptor surfaces for coregulator interaction. This concept was tested with the use of combinatorial phage display to identify peptides that interact with different surfaces on the ERs. Initial studies demonstrated that ER-binding peptides could function as sensitive conformational probes, capable of distinguishing between ER subtype, and apo-receptor vs agonist- or antagonist-bound receptor (63). Further analyses revealed that these peptides could even distinguish between 17β-estradiol and its metabolites estrone and estriol, and furthermore, could differentiate among structurally related antiestrogens such as the SERMs tamoxifen, nafoxidene, and idoxifene (Figure 3⇓) (63, 73). The screening of focused combinatorial phage libraries expressing peptides containing consensus coactivator (LXXLL) and corepressor (CoRNR box) sequences for NR binding led to the identification of probes that could clearly differentiate between ligands, ER subtype and DNA context (21, 43, 63, 68, 74, 75). This provided compelling evidence that these reagents were detecting cofactor interaction surfaces on the ERs. Thus, with this new technology it became increasing apparent that 1) ligands known to produce distinct biological effects induce different conformational changes in the ERs, and 2) a strong correlation exists between receptor conformation, coregulator recruitment, and biological activity.
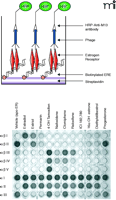
Different ligands induce distinct structural alterations in ERα. A. A drawing indicating the various components of an enzyme-linked immunosorbent assay (ELISA) used to identify distinct changes in ER structure as mediated by binding to peptides displayed on the surface of phage. B. Typical results from a ER-phage ELISA utilizing ERE-containing oligonucleotides. Purified ERα was added in saturating concentrations and then incubated with differing ligands, including the agonist 17β-estradiol and its metabolites estriol and 16α-OH estrone; the ER pure antagonist ICI 182,780; the selective estrogen receptor modulators (SERMs) tamoxifen, nafoxidene, raloxifene; the pharmaceutical agents premarin, clomiphene, and diethylstilbestrol; and solvent and progesterone as negative controls (functionally equivalent to apo-ERα). Individual phage clones, each expressing specific peptides (α/β I, α/βII, α/β III, α/β IV, α/β V, α I, α II, α III) were incubated with the (oligo)DNA-receptor-ligand complexes, unbound phage were washed away and bound phage were detected with an M13 antibody conjugated to HRP. Assays were developed with 2,2-azinobis(3-ethylbenzothiazoline)-6 sulfonic acid and hydrogen peroxide for 10 min and stopped by the addition of 1% SDS. The visual results of the colorimetric assay are shown [Figure adapted from (63)].
A recent report profiling ligands based on ER-peptide binding characteristics demonstrated that receptor conformation is a better indicator of the agonist activity of different ER ligands than cell-based reporter gene assays (76). This observation and those from other studies cited above indicate the promising potential use of ER-peptide profiling assays to identify novel ER pharmaceuticals. It is also possible to envision a use for these peptides in identification of environmental agents with endocrine-disrupting activities. This idea stemmed from the finding that different environmental estrogens, when complexed with either ER subtype, demonstrate significantly different binding preferences for a series of LXXLL-containing peptides (43); this is consistent with the ability of these compounds to display distinct transcriptional agonist activities on the ERs in mammalian cells. Collectively, the identification of ER-binding peptides has demonstrated that there exists a strong correlation between receptor conformation and biological activity and has indicated the potential use of these peptides as tools to screen for mechanistically distinct ER pharmaceuticals and toxicological agents.
Development of Mechanistically Distinct ER Antagonists that Target ER-Cofactor Interactions
All of the known ER antagonists function by binding to the ER ligand-binding pocket, thereby occluding agonist access and inducing a conformational change in the receptor that prevents efficient interaction with transcriptional coactivators. A more direct method of antagonizing ER function, however, might be achieved downstream of ligand binding through direct interference with receptor-coactivator interactions. To address this possibility, our laboratory successfully used combinatorial phage library screening to identify LXXLL peptides that bind with high affinity to the coactivator pockets of the ERs. By introducing these peptides into mammalian cells it was possible to effectively inhibit ERα and ERβ transcriptional activities by disrupting interactions between the receptors and cellular coactivators (21, 74). Given the structural conservation among NRs, it was not anticipated that it would be possible to identify peptides specific for the ERs over other receptors; however, by altering the sequences flanking the LXXLL motifs, peptides were identified that function as highly specific antagonists of ERα or ERβ and not other NRs.
ERα is overexpressed in the majority of breast cancers, and ERβ is increased in tumors that have developed tamoxifen resistance (66, 77). Thus, there is an unmet medical need to develop novel antiestrogens as breast cancer therapeutics and tools to define the role of the ERs in breast cancer cell biology. The identification of these peptide reagents afforded us the opportunity to speculate on the potential therapeutic use of peptidomimetic derivatives as ER antagonists. Given that the peptides display no detectable interaction with antagonist-bound receptor (21, 74), it is possible that coadministration of suitably formulated ER peptide antagonists with tamoxifen could provide a mode to completely block estrogen-stimulated proliferative pathways in the breast. Recent evidence suggests that tamoxifen resistance may occur through increased expression of coactivator proteins, which may permit cells to recognize tamoxifen as an ER agonist and stimulate growth (59, 78). The identification of peptide antagonists that disrupt the ER-coactivator interaction provides a new mode for blocking the proliferative effects of estrogens in both antiestrogen-sensitive and -resistant breast cancer cells. Furthermore, these peptide antagonists or peptidomimetic derivatives could possibly be developed as second-line therapeutics for ER-positive, tamoxifen-refractory breast tumors.
Summary
In the past decade it has become clear that the sole existence of two forms of the ERs is not sufficient to account for the diverse biological roles of estrogens and pharmacological activities of synthetic ER ligands. Recruitment of coregulatory complexes enables the ERs to communicate with the general transcription apparatus, possess the catalytic activities required for chromatin modification, and capacity to integrate extracellular signals and translate them into transcriptional and biological events. Cofactors provide a mode for achieving specificity in target gene expression and provide for the ligand- and tissue-selective activities of the ERs. The strong association between coregulator levels and ER function in estrogen-associated pathologies continues to prompt many efforts in the field to define the role of these factors in disease and develop pharmaceuticals with unique mechanisms for modulating ER activity in a clinical context. It is expected that this knowledge will provide for future development of novel ER-targeted therapeutic agents by 1) identifying ligands that induce unique ER conformations and ER-cofactor complexes, and 2) developing agents that act directly upon existing ER-cofactor complexes.
Acknowledgments
The authors would like to thank our colleagues for their outstanding contributions to the nuclear receptor-cofactor field. We regret that due to space constraints we were unable to include several salient studies by many groups.
- © American Society for Pharmacology and Experimental Theraputics 2005
References

Donald P. McDonnell, PhD, is the Glaxo-Wellcome Professor of Molecular Cancer Biology in the Department of Pharmacology and Cancer Biology at Duke University Medical Center in Durham, NC. In addition, he serves as the director of the departmental graduate studies program. Dr. McDonnell came to Duke in 1994 from Ligand Pharmaceuticals, Inc. where he was the director and head of molecular biology. His work has focused in recent years on the genetic and pharmacological dissection of the steroid hormone receptor signal transduction pathways. The insights from this work have led to the discovery and development of novel estrogen and progesterone modulators that are being evaluated clinically as treatments for cancer and osteoporosis. Please address correspondence to DPM. E-mail:donald. mcdonnell{at}duke.edu; fax: (919) 684-2212.

Julie M. Hall, PhD, is a Research Scientist in the Department of Pharmacology and Cancer Biology at Duke University Medical Center in Durham, NC. She received a PhD in Pharmacology from Duke in 2000, and completed a three-year postdoctoral fellowship at the National Institute of Environmental Health Sciences, NIH. Her research interest in both of these positions was the molecular mechanisms of estrogen receptor action. Since returning to Duke two years ago as a Junior Faculty member, her research has continued to focus on the molecular pharmacology of nuclear receptor action.

