Antibody Conjugates and Therapeutic Strategies
Abstract
Immunotherapeutics represent the largest group of molecules currently in development as new drug entities. These versatile molecules are being investigated for the treatment of a range of pathological conditions including cancer, infectious and inflammatory diseases. Antibodies can be used to exert biological effects themselves or as delivery agents of conjugated drug molecules. Site-specific delivery of therapeutic agents has been an ultimate goal of the pharmaceutical industry in order to maximize drug action and minimize side effects. Antibodies have the potential to realize this objective and in this review we will examine some of the main strategies currently being employed for the development of these diverse therapeutic molecules.

Introduction
Antibodies represent the single largest class of new drug entities under development at this time. There are at least twenty immunotherapeutics currently marketed, with some 150 developmental products currently in clinical evaluation (1). The possibility of using antibodies as therapeutics was considered almost as early as immunologists came to understand their role as our body’s natural defense against foreign agents. This potential, however, was only initially realized with the development of monoclonal antibodies (MAbs) in the 1970s (2). Immortalization of murine antibody-secreting B cells by hybridoma technology and their continual cell culture permitted large scale production of a single, pure antibody species, a prerequisite for therapeutic development. Initial attempts to develop antibody-based therapeutics encountered difficulties when it became clear that the human body recognized the murine MAbs as foreign molecules and developed an antibody-mediated immune response to clear these from the body (3). The development of these human anti-murine antibodies (HAMA) seriously reduced the half-life of the therapeutic MAbs in the blood stream, particularly with prolonged administration, rendering them useless as drug strategies.
In order to reduce the HAMA response to administered MAbs, protein engineers manipulated the nucleotide sequences of the antibodies to convert murine-like antibodies to more human-like antibodies. Techniques now used routinely for the “demurinization” of MAb include complementarity-determining region (CDR, the region of an antibody that is most involved in binding an antigen) grafting, humanization, and even the production of transgenic mice that make fully human antibodies. These advances in antibody engineering have created a surge in development of antibody-based therapeutic agents such that within the global pharmaceutical industry, larger companies, once slow to embrace antibody technology, have moved to secure intellectual property (IP). The securing of IP has been achieved by large licensing deals, takeovers, and mergers with biotechnology companies who offer novel antibody technology platforms or promising lead antibody drug candidates (4).
Antibody therapeutics exert their biological effects by a number of different mechanisms. Two possible mechanisms operate through the involvement of the immune system to induce cytotoxicity of the target cell population. The first of these is antibody-dependent cellular cytotoxicity (ADCC), where immune effector cells are recruited to the disease by virtue of the antibody Fc chain site once the antibody has bound to its target (5); however, the relevance of this mechanism, at least in clinical oncology, remains controversial (6). Alternatively, the Fc portion of a therapeutic antibody, as in the case of the CD20-specific rituximab (7), can recruit the activation of the complement cascade, resulting in the formation of the membrane attack complex, a mechanism referred to as complement-dependent cytotoxicity (CDC). Antibodies can also be used to modulate signaling pathways within the targeted cells. Bevacizmab (Avastin®) blocks angiogenesis by binding and sequestering soluble vascular endothelial growth factor (VEGF), and trastuzumab (Herceptin®) binds to the cell surface protein HER2/neu, inhibiting proliferation pathways that are transduced through this signaling receptor.
In addition to these mechanisms of utilizing the antibody as the effector molecule, increasing attention has turned to the possibilities of using antibodies as delivery conduits and as a means to targeting disease sites specifically. By selectively targeting the drug to the site of disease, an antibody can induce the desired biological effects with improved therapeutic index. To date, three antibody conjugate therapies have been approved for marketing, namely, the radioimmunoconjugates ibritumomab tiuxetan (Zevalin®) and [131I]-tositumomab (Bexxar®) for the treatment of lymphoma and the drug conjugate gemtuzumab ozogamicin (Mylotarg®) for the treatment of acute myeloid leukemia. In this review, the different strategies employed in the production and application of antibody conjugates are discussed.
Antibody Conjugates: Theory and Application
Effective delivery to the site of disease is a prerequisite for high efficacy and low toxicity of any drug substance. It is clear that antibodies can participate in this context by facilitating the transport of a drug cargo within the body and thereby invoking the often cited “magic bullet” concept, as put forward by Ehrlich over a century ago. Conjugation of a drug to an antibody makes it possible to achieve excellent localization of the drug at the desired site within the body (8). This increases the effective drug concentration within this target area, thereby optimizing the therapeutic effect of the agent. Furthermore, with targeted delivery, the clinician may be able to lower the dose of the therapeutic agent––something that is particularly relevant if the drug payload has associated toxicities or if it is to be used in the treatment of chronic conditions.
There are several basic considerations to be addressed when designing and applying an antibody conjugate to a particular disease model. The choice of drug payload is critical to ensure desired efficacy towards the targeted disease and that the stoichiometry, orientation, or associated chemistry of conjugation to the antibody will not hinder its biological activity. Furthermore, the design of any immunotherapeutic agent centers on the selection of antigens or biomarkers that are specific and accessible to antibody binding at the disease site. Not surprisingly, biomarker identification and characterization is a key focus for pharmaceutical and biotechnology companies alike and relies heavily on technologies such as DNA microarrays and proteomic techniques. Consequently, companies rapidly secure IP around newly identified biomarkers that could possibly lead to antibody-based drugs.
Despite substantial investment over the last ten years in biomarker identification, the number of therapeutically exploitable disease biomarkers that have been discovered is somewhat disappointing. This has led to renewed thinking as to how to examine novel strategies that can maximally exploit this small group of biomarkers, particularly when the biomarker is not a drug target. Antibody-drug conjugates are an ideal way to utilize these identified biomarkers that cannot be targeted therapeutically by naked antibodies alone. We discuss below the main conjugate classes under investigation, together with those that have the potential to emerge as effective therapeutic agents against cancer and a wide range of other disease conditions.
Peptide and Protein Conjugates
Numerous peptides and proteins elicit desirable therapeutic effects, but frequently utilization in therapeutic approaches have met with limited success owing to possible toxicity in the systemic circulation, poor bioavailability, degradation, and an inability to achieve useful therapeutic measures. Conjugation of these proteins to a suitable antibody or antibody fragment can circumvent these problems, achieving greater stability and higher specificity in delivery to the site of disease (9).
Peptides and proteins can be attached to each other by either chemical or recombinant means. The earliest examples of enzyme-antibody conjugates used chemical cross-linkers, such as maleimide, to couple thiol moieties between cysteine residues on each partner (10). This nonspecific approach can result in multiple products, with one or both partners inactivated by the coupling reaction. Alternative methods involve the specific conjugation of particular residues in the protein species, such as the prior oxidation of N-terminal threonine residues to produce a reactive aldehyde group that could then be specifically modified using cross-linking reagents (11). Currently, however, the most popular methodology uses recombinant DNA techniques to fuse the antibody open-reading frame to that of the therapeutic protein, giving rise to an antibody-protein fusion after translation. This approach to conjugation is much more amendable to the manufacture of a uniform protein species for therapeutic application (12). Frequently, the optimal fusion of protein entities can be a case of trial and error, often necessitating the incorporation of a short flexible linker to ensure maintenance of the two distinct biological activities. When full immunoglobulin G (IgG) antibodies are used, the most common strategy is the attachment of the cytokine at the C terminus of the antibody heavy chain (13), whereby with single chain antibody fragments (scFv), successful fusions have been achieved at both the N and C termini.
A particular antibody–therapeutic protein strategy that has been investigated by several groups is the selective activation of anticancer prodrugs by enzymes conjugated to a delivery antibody, frequently referred to as antibody-directed enzyme prodrug therapy (ADEPT) (14). This strategy has been investigated in the treatment of solid tumors, whereby an antibody-enzyme conjugate is administered systemically, where it clears from the circulation and localizes to its target by virtue of the antibody binding to its specific biomarker on the tumor. Subsequently, a suitable non-toxic prodrug is then administered, which can be converted to its active cytotoxic form by the enzyme attached to the antibody. The activated drug penetrates the tumor cells and exerts its lethal effect in a localized manner (Figure 1⇓). The ADEPT strategy facilitates the specific targeting of the disease site and directs toxicity to the target cell population. Crucially, this approach has the ability to target adjacent diseased cells not directly labelled by the antibody-enzyme conjugate and permits penetration of the free drug into the tumor mass. This effect is lacking during naked antibody or antibody-toxin conjugate solid tumor therapeutics, which can be viewed as a potential drawback to these other approaches.
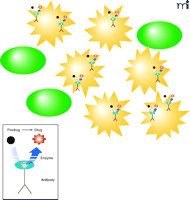
Basis of ADEPT strategies. The antibody-enzyme conjugate specifically binds to a tumor-specific marker on tumor cells (yellow) and not on normal cells (green). Upon subsequent systemic application of the prodrug, this is converted to active cytotoxic agent at the site of antibody binding. The cytotoxic agent exerts is biological effect within the vicinity of antibody binding.
In the design of an ADEPT approach, several factors must be considered. First, the choice of a suitably specific antibody and its cognate antigen are paramount. The chosen antigen is normally on the tumor surface itself, but can also be on surrounding tissue in some instances. An exquisite example of this is the targeting of the extra domain-B (ED-B) of fibronectin by L19, a recombinant antibody fragment (15). ED-B is specifically expressed in large amounts on neovasculature as a result of strongly pro-apoptotic stimuli secreted by many aggressive solid tumors. L19 localizes at these sites in a number of tumor models and has been used to deliver a wide range of therapeutic conjugates. The second consideration is the choice of enzyme to be attached to the antibody species. To add specificity to the activation of the prodrug, it would be pertinent to select a catalytic activity that would not be found at the target site––for example, an activity originating from a foreign organism, such as bacterial β-lactamase or carboxypeptidase G2. Alternatively, mammalian enzymes that are unlikely to be found in high basal concentrations in the body, such as carboxypeptidase A and β-glucuronidase, have also been used. Both strategies can ensure effective activation of the prodrug but are not without their drawbacks. Although high specificity can be obtained with bacterial or fungal enzymes, severe immunogenicity towards these foreign proteins is an important concern. Problems of immunogenicity may be avoided by using mammalian enzymes; however, because similar (mammalian) catalytic activities are likely to be present in the patient, the specificity of prodrug activation could be confounded by activation at sites other than the intended target region. The final component of an ADEPT approach is the actual prodrug itself, which must be compatible substrate of the activating enzyme. Catalytic activation of prodrugs has long been a major focus of pharmaceutical research (16). Methotrexate prodrugs activated by carboxypeptidase A, etoposide phosphates by alkaline phosphatase and acid mustard compounds by carboxypeptidase G2 are just a few of the more common examples. Given the plethora of enzymes that occur in nature and the numerous choices of potential prodrug formats, a broad range of ADEPT strategies have been investigated (Table 1⇓).
Selected Enzymes and Pro-drug Combinations Used in ADEPT Strategies
Enzymes are not the only protein-based conjugates that have been investigated as immunoconjugates. Cytokines and other small proteins that can elicit a strong biological response are attractive as therapeutic agents. Attachment of such molecules to antibodies can improve their stability and plasma half-lives and facilitate accurate delivery. These improved parameters are particularly important given the exceptionally strong biological effects these proteins can elicit and the risk of possible unwanted side-effects following systemic delivery. Antibody-cytokine conjugates, or immunocytokines, have attracted particular interest for tumor treatment because of their potential to cause localized stimulation and activation of immune effector cells. Tumor necrosis factor–α (TNF-α), granulocyte-macrophage colony stimulating factor (GM-GSF), interferon γ (IFNγ) and interleukin-2 (IL-2) are just a few of the cytokines that have been investigated in immunocytokine approaches (17). Reisfeld and coworkers have focused on IL-2, a pleiotropic cytokine that stimulates the activation of T-cells, natural killer (NK) cells, and macrophages, to enhance these cells recognition and destruction of tumor cells. From as early as the 1980s, the potency of IL-2 was appreciated, but with systemic delivery, an exploitable therapeutic index was not achievable. To localize delivery of the cytokine, this group fused IL-2 to an antibody directed towards disialoganglioside GD2, a ganglioside that is over-produced in melanomas. In animal models, this immunocytokine afforded protection against the development of lung and hepatic metastases from injected melanomas (18). Further study confirmed that the protection mechanism was reliant on activation and recruitment of T cells to the tumor site. It has been postulated that IL-2 provides a co-stimulatory signal for the activation of CD8+ T cells which, in combination with signals elicited by antigenic peptides displayed on the MHC-I molecules of the tumor cells, are able to proliferate and eradicate the tumor. The humanized version of this antibody–IL-2 conjugate is currently in clinical trials for treatment of melanoma (19).
TNF-α is toxic to tumor cells, exerting its activity on endothelial cells of the tumor neovasculature. As is the case with IL-2, systemic delivery of TNF-α is not possible owing to its high toxicity to normal tissue. Neri and coworkers have conjugated TNF-α to their L19 fibronectin-specific antibody and demonstrated therapeutic indices unattainable with unconjugated cytokine. The anti-tumor activity of this experimental therapeutic agent is enhanced synergistically when used in combination with an L19–IL-2 fusion (20). Both these molecules are now in early stage clinical evaluation.
ADEPT and immunocytokines are only two of a range of immunotherapeutic protein or peptide strategies currently in pre-clinical and clinical development for a range of diseases, predominantly in the field of oncology. Any peptide or protein shown to have therapeutic effect can be exploited potentially in the form of an immunoconjugate for the treatment of a plethora of disease conditions. The range of possibilities will ensure continued interest in this area and marketing of eventual drugs in this class.
Drug-Immunoconjugates
Conventional cancer treatment frequently involves administering a regime of chemotherapeutic drugs that may be used as the primary means of intervention or as an adjuvant to surgical removal of the tumor mass. Systemic administration of chemotherapeutic drugs results in the death of actively dividing cells within the body, specifically cancer cells, but also has the undesired side effects of destroying oral and intestinal mucosa, hair follicles, and bone marrow. Much attention has been directed to selective targeting of these agents by conjugation to antibodies against tumor specific markers, in an attempt to improve efficacy and reduce side effects.
As early as the mid-1970s, antibody-drug conjugates were being investigated as an approach for targeting chemotherapeutic drugs to tumor cells (21). In these early investigations, drugs such as methotrexate were covalently linked to human serum albumin as a carrier, which in turn could then be chemically linked to a monoclonal antibody. Garnett and coworkers compared the use of this approach for targeted cytotoxicity with that of conventional systemic delivery toward an osteogenic sarcoma cell line. The conjugate was selective in its action and was preferentially cytotoxic towards antibody-reactive cell types under a competitive assay with non-conjugated antibody (22).
In 2000, the FDA granted approval for the first antibody drug conjugate for human therapeutic use. Humanized CD33-specific MAb conjugate [gemtuzumab ozogamicin (Mylotarg®)] was licensed for the treatment of acute myeloid leukemia (AML) in patients over sixty years of age and deemed unsuitable for other chemotherapy (23). This “first-in-class” therapeutic consists of gemtuzumab, a labile linker attached to Lys side-chains, and then conjugated to a calicheamicin hydrazide derivative. Approximately two or three molecules of calicheamicin are attached per antibody, and the therapeutic formulation consists of 50% of the antibody labeled by the drug. Mylotarg® binds to the cell surface marker CD33, a receptor overexpressed on acute myeloid leukemia cells and on normal leukocytes and leukemic progenitors, but which is absent on normal stem cells. Once Mylotarg® binds to CD33, the receptor-antibody conjugate complex is internalized and hydrolyzed. The resulting hydrolysis releases calicheamicin, which freely localizes in the nucleus, initiating DNA alkylation and promoting cell death (24, 25). In preclinical models, Mylotarg® selectively and potently inhibited CD33+ AML cell lines, and clinical studies have revealed complete remission in up to 30% of adults with relapsed CD33+ AML (26).
In addition to the selection of a suitable chemotherapeutic agent and exploitation of a highly specific antibody towards a biomarker of myeloid leukemia, the efficacy of Mylotarg® is, in large part, because of the linker chemistry between antibody and drug. The linker region is crucial in these molecules to ensure that the hybrid molecule: 1) is stable, 2) can successfully deliver the drug to the target cell, tissue or organ, and 3) can present the drug appropriately to permit therapeutic action. The most popular approach for the development of immunochemotherapeutics is to use an antibody that upon binding to its specific cell surface biomarker will internalize; a mechanism referred to as receptor-mediated endocytosis. Endocytosis will direct the immunochemotherapeutic agent to endosomes, where fusion with activated lysosomes initiates metabolism and degradation of the endosome contents. The nature and composition of the activated lysosomes offer many possibilities to the drug developers for the specific release of the drug compound. In the case of Mylotarg®, a hybrid linker is used that contains both a hydrazone and sterically-confined disulfide, which are labile only in acid (hydrazone) and reductive (disulfide) environments. In the activated lysosomes, the acidic conditions (pH 5) and high glutathione concentrations can cleave the hydrazone and reduce the disulfide, resulting in delayed release of the drug from its delivery antibody after endocytosis. In addition to the hydrazone and disulfide linkers (as used in Mylotarg®), other linker chemistries are being examined. One of the most promising strategies involves the used of protease-specific peptide sequences, which can be cleaved by activated proteases, such as the lysosomal cysteine protease cathepsin B (27). Depending on the nature of the drug or toxin, protease recognition sequences can be created through genetic engineering or by peptide chemistry. Using this strategy, Dubowchik and colleagues created protease-sensitive sequences in conjugates and demonstrated improved stability and specificity as compared to those observed in hydrazone conjugates (28). In these studies, the investigators found that dipeptides sequences that are sensitive to proteolytic cleavage by cathepsin B (e.g., phenylalanine-lysine and valine-citrulline) were effectively hydrolyzed by the protease when used as a linker between antibody-doxorubicin conjugate, after internalization of the conjugate-antigen complex.
In addition to calicheamicin, doxorubicin represents one of the most commonly researched drugs in antibody-based applications. Doxorubicin, an anthracycline, produces its cytotoxic effects by alkylation of double-stranded DNA, resulting in interstrand cross-linking, triggering apoptosis (29). Tolcher and coworkers conjugated this chemotherapeutic agent (via a hydrazone linker) to a chimeric antibody (BR96) that targets Lewis(y), a tetrasaccharide overexpressed on the surface of many tumors, and in 75% of breast carcinomas (30). Most normal tissues, except for epithelial cells in the GI tract, do not express this tumor antigen (31). Although BR96-doxorubicin conjugate demonstrated efficacy against metastatic breast and non-small cell lung tumors, Phase I and II clinical trials also highlighted significant gastro-intestinal toxicity due to the presence of the antigen in the GI tract; it is now being evaluated under reduced dosing in combination therapy with docetaxel (32).
A further drug family that has been used in antibody-targeted delivery are the auristatins, based on the naturally occurring peptide termed Dolastatin 10. The auristatins are highly potent peptides that inhibit tubulin polymerization, producing antimitotic effects that can be exploited therapeutically (33). Synthetic auristatin analogs such as monomethyl auristatin (MMAE) and auristatin E (AE) have been conjugated to tumor-specific antibodies. Senter and colleagues succeeded in producing highly efficacious molecules that targeted carcinomas and hematologic malignancies [using Lewis(y)- and CD20-specific antibodies, respectively] and were effective at one-sixtieth of the maximum tolerated dose (34).
In addition to these chemotherapeutic agents, protein-based toxins have also been examined in their potential as immunotherapeutics. Using recombinant antibody technology it is possible to express antibodies or antibody fragments with a toxic fusion protein partner that can be cleaved upon internalization by proteases such as cathepsin B or E. Protein toxins that have been conjugated or fused to delivery antibodies include ricin, gelonin, Pseudomonas exotoxin, diphtheria toxin, and RNase (35). Gelonin consists of a single polypeptide and is a ribosome-inactivating toxin with a biological activity similar to that of ricin, a potent inhibitor of protein synthesis. The humanized CD33-specific MAb HuM195 linked to recombinant gelonin exhibits much more potency than the non-fused MAb when applied administered in vitro to primary blast cultures from patients with AML (36). Development of this and other immunotoxins as effective therapeutics represents a promising area of research, especially when one considers the wide range of toxic proteins or molecules that can be conjugated to a targeting antibody. The main challenges for this type of immunoconjugate are preventing or limiting toxic side effects and rapid clearance problems owing to the inherent immunogenicity of the toxins, which are almost entirely nonhuman in nature.
Radioimmunoconjugates
The use of nuclear medicine, both in diagnostics and therapeutics relies implicitly on accurate delivery to the disease location. Within this field, the application of antibodies as delivery agents for the radioisotopes holds much promise, as two of the three currently licensed antibody conjugates at this time are radioimmunoconjugates. As with other immunoconjugate strategies, confirmation that a radioisotope can be stably conjugated to an antibody without adversely affecting its biological activity has led to their widespread evaluation in both the imaging (immunoscintigraphy) and radioimmunotherapy (RIT). Depending on the nature of the radioligand used, the same antibody can be used to target specifically the site of disease for either toxic effector function, or imaging and diagnosis.
The FDA has approved two radiolabeled antibodies––ibritumomab tiuxetan and tositumomab––both of which target CD20, a cell surface marker expressed on over 95% of B cells and B cell lymphoma. CD20 is not expressed on stem cells or on terminal differentiated plasma cells; therefore, the removal of CD20+ cells from the patient has no long-term effects. CD20-targeted RIT has been successful because lymphoma cells are very accessible and allow rapid binding of MAb. The disease characteristics also allow delivery of high energy particles to the tumor cells.
The 90Y-conjugated ibritumomab tiuxetan (Zevalin®) relies on the covalent conjugation of a metal chelating agent, tiuxetan, to a chimeric IgG1 kappa antibody (37–39). The tiuxetan group chelates the 90Y metal isotope for use as a therapy or, alternatively, the gamma-emitter 111In for use in imaging. Tositumomab (Bexxar®), consists of a CD20-specific antibody conjugated to 131I, using direct oxidant-based chemistry, before chromatographic purification (40).
Both approved antibodies (Bexxar® and Zevalin®) employ beta-emitting radioisotopes that have therapeutic applications in the treatment of hematological malignancies. Beta emitters induce cell death by forming free radicals in the target cell. The beta emissions of 90Y, with a half-life of sixty-four hours (Table 2⇓), deposit energy over a relatively large area (i.e., several mm), leading to possible dissipation into neighboring cells but less likely to cause widespread damage to surrounding normal tissue. Novel radiolabels, however, with more favorable linear energy transfer, half-life decay, and path length characteristics are in development. Alpha emitters (i.e., 211At) are presently being considered for therapeutic use because of their high ionization potential per unit path length. They are more effective at selectively killing target cells and lack the ability to penetrate adjacent cells (41). The careful selection and matching of differing energy-depositing radionuclides to disease lesions, depending on type and bulk of tumor tissue, will facilitate improved efficacies to these radioimmunoconjugates (42).
Common Alpha and Beta Radionuclides Used for Radioimmunotherapy
The challenges faced by radioisotope delivery are similar to those faced by other modes of immunotherapy, namely specific localization, clearance and system toxicity. One methodology being examined is a pre-targeting approach using bispecific antibodies (43). The development of recombinant antibodies with dual specificity (bispecific) toward a tumor-associated target and toward a functional moiety such as a metal chelating group (for radionuclide binding) has introduced the concept of “pretargeting” for certain diseases. In this therapeutic approach, the patient is given a suitable bispecific antibody, which is given sufficient time to localize to the disease site, while the excess remainder of the antibody is cleared. The systemic application of a suitable radionuclide hapten follows, which then binds to the other binding site on the antibody, thereby allowing localization of the radionuclide to the site of disease to exert its cytotoxic response (44). Other pretargeting approaches have exploited streptavidin-biotin chemistry (45) and complementary oligonucleotide approaches (46) in order to home the targeted radionuclide to the site of bispecific antibody binding. The pretargeting approach allows improved flexibility in the nature of the radionuclide used for both therapy and imaging. Recent advances in availability of various radionuclides and in antibody engineering abilities have injected interest and optimism in this, and other radioimmunoconjugate methodologies.
The use of radiolabeled recombinant antibody fragments has also aided the specific targeting of tumors with more favorable clearance profiles and less non specific binding, for example by attenuation of Fc-FcRn (neonatal Fc receptor) interactions (47). Biodistribution studies have shown that engineered antibody fragments such as single chain Fv (scFvs), diabodies, and minibodies have superior uptake from the blood, particularly useful for imaging (48) and, in some cases, they exhibit favorable tumor localization and penetration (49) (Box 1) .
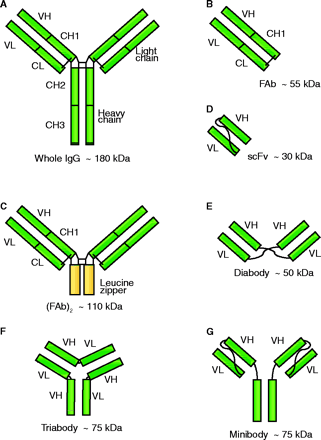
Recombinant Antibody Fragments
The IgG antibody (A) consists of a heavy and a light chain (H and L), both of which contain variable (V) and constant regions (C). The binding pocket of the antibody that recognizes its specific epitope consists of the variable regions of both the heavy and light chains (VH and VL), and only this region is required to produce a molecule capable of binding.
FAb fragments can be produced by proteolytic cleavage of a monoclonal antibody, or by recombinant engineering, to remove the Fc domain, and can be of the form FAb or (FAb)2. The FAb fragments include the complete light chain of the antibody linked by a disulphide bond to the VH-CH1 fragment of the heavy chain (B). The production of homospecific or bispecific (FAb)2 dimeric antibodies is possible, and dimer association can be strengthened by inclusion of a genetically engineered leucine zipper (C).
By using recombinant technology, it is possible to generate a variety of smaller fragments of the IgG molecule that retain or have improved binding affinities. One of the most common forms is the single chain variable fragment (scFv), produced by combination of VH and VL joined by a flexible peptide linker (D). In the scFv molecule, the VL and VH domains can freely associate in an intramolecular relationship. Shortening of the linker length can inhibit this intramolecular interaction, and intermolecular association of VL and VH domains becomes more energetically favorable, resulting in the formation of diabodies (E), triabodies (F) or even tetrabodies. The scFv form has been further modified to include constant domains such as CH3 for the creation of minibodies (G), which are composed of either homo- or heterodimers of the scFv-CH3. Minibodies have the advantage of increased stability and bivalent specificities if required.
Conventional whole IgG radiolabeled MAbs exhibit impaired killing efficacy arising from the short retention time of the radionuclide within the targeted cell. The problem of retention time within target cells can be addressed by “residualizing” the radiolabels. The principle of the technique involves the inclusion of a radionuclide as a component of a non-metabolizable peptide (for example, diethyl-enetriaminepentaacetic acid–appended radioiodinated peptides that contain D-amino acids) designed to become internalized by receptor-mediated endocyotsis and localized in the lysosome following antibody catabolism (50). Once trapped, the radiolabel is capable of causing maximum effect.
As with all antibody-based therapeutic approaches, the development of accompanying diagnostic tools to the radioimmunoconjugates is as important as the therapeutic agent itself. In this area, the emerging techniques for the application of radiolabeled antibodies in whole-body or tumor imaging is particularly exciting, providing data on tumor localization and metastases as well as insight into biodistribution patterns of potential medications. Positron emission tomography (PET) is the four dimensional imaging of a radiotracer (e.g., 18F, 64Cu, 86Y, 76Br, or 124I) within a living system (51). The diagnostic potential of positron emitter–coupled antibodies has suffered because of the short half-life of radiotracers and the slow systemic clearance of antibodies. Recent evidence has demonstrated that the positron emitter 89Zr, which has a kinetic profile that matches that of whole antibodies, can act as a reliable predictor of therapeutic antibody biodistribution (52). Advances in antibody engineering to improve the clearance and localization kinetics of the labeled antibodies will further enhance their use, and the use of more stable conjugation chemistries [for example, sulfosuccinimidyl-3-(4-hydroxyphenyl) propionate)] will contribute to the growing application and success of radioimmunoconjugates.
Immunocolloidal Conjugates
The application of colloids, such as nanoparticles and liposomes, has become an important focus within drug delivery and targeting. The small size of colloids allows for their passage through the narrowest of capillaries and endothelial spaces, delivering a wide range of drugs to the body for sustained periods of time. In addition, they form a drug delivery vector with much adaptability, making them a generic system that can be tailored to carry several types of drug substance and have a conjugated targeting system decorated upon their outer surface.
Targeted drug delivery using colloidal systems can be achieved by either passive or active targeting. The former refers to the accumulation of a carrier system at distinct sites dictated by factors of physicochemical or physiological means (53). This is exemplified by selective nanoparticulate uptake at sites of inflamed colonic mucosa. Similarly, the enhanced permeation and retention effect permits colloidal nanoparticles to accumulate in tumor tissue compartments (54), which is driven by the vascular endothelium of angiogenic blood vessels having gaps (600–800 nm) between adjacent endothelial cells that exceed the nanoparticle diameter (100–500 nm). Also, nanoparticles with hydrophobic surfaces demonstrate increased uptake by the mononuclear phagocytic system and may be a useful strategy to treat diseases of reticuloendothelial organs. Active targeting, on the other hand, requires modification of the naked nanoparticle, usually by conjugation with a recognizing moiety, such as a specific and selective antibody to facilitate targeting (55).
To achieve antibody-mediated targeting, peripheral attachment of the antibody to the colloid is needed. This is generally a two-step procedure where the nanoparticle surface is primed with a suitable functional group prior to the second step involving ligand attachment. One method of nanoparticle priming can be achieved using a formulation with modified polymeric strands (Figure 2A⇓), such as those based on poly(lactide-co-glycolide) acid (PLGA). The use of modified strands ensures that the resulting copolymer retains some biodegradability and biocompatibility, a property that is conferred to the primed nanoparticle. Amine functionality, for example, can be added by copolymerization with poly(lysine) (56) or by using newly synthesized polymers, such as PEG-NH2 (Figure 3⇓). Further modification is possible, where block copolymers comprising poly(lactic acid)-poly(ethylene glycol)-biotin
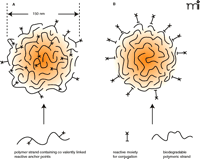
A schematic representation of polymeric nanoparticles of approximately 150 nm diameter with anchor points for conjugation. These anchors can form an integral and covalent part of the polymer stand prior to nanoparticle assembly (A). PLGA, commonly used to make solid nanoparticles, lacks the necessary functional groups for covalent conjugation. Functionality added prior to assembly ensures that at least some of the functional groups exist at the surface, with most submerged within the nanoparticle matrix. Alternatively, functionality can be adsorbed onto the nanoparticle surface once its assembly is complete (B).
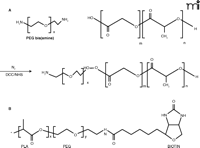
Chemical structures of molecules used in linking antibodies to active group moieties. A. Copolymerization of poly(ethylene glycol)-bis(amine) with poly(lactide-co-glycolide) to give a polymeric strand presenting a terminal end amine group. B. Further attachment of a biotin group gives a polymeric strucure that can be formed into nanoparticles that presents an attachment point for avidin-containing ligands.
(PLA-PEG-biotin) have been described and used to assemble nanoparticulate systems (57) (Figure 3B⇑). This modification exploits avidin-biotin binding and is a novel method for noncovalent bonding of an avidin-containing ligand to a biotin-presenting nanoparticulate surface. A simpler method for introducing functionality to a nanoparticle periphery is to use surface adsorption without any form of covalent chemistry (Figure 2B⇑). Most nanoparticle suspensions require a surface-bound stabilizer such as a nonionic surfactant to prevent coalescence. Replacing some of this stabilizer with poly(ethylene-alt-maleic acid) has been used to introduce carboxylic groupings to the nanoparticle surface (58). Similarly, adsorption of avidin-palmitic acid has been used to generate an avidin-rich surface. These approaches may suffer drawbacks, such as difficulty in preparation and loss of the targeting effect that is exacerbated with time owing to desorption or degradation of adsorbed groups as the nanoparticle scaffold erodes.
Liposomes are colloidal drug carriers, of similar dimension to nanoparticles, but consisting instead of one or more phospholipid bilayers enclosing an aqueous core. They take on the form of large multilamellar (MLV), small unilamellar (SUV), or large unilamellar (LUV) vesicles. Water-soluble drugs occupy the aqueous compartments, whereas those of more lipophilic character occupy the lipid bilayers. Drug targeting with liposomes has been discussed in depth elsewhere, whereas the term “immunoliposome” has been coined to describe a liposome conjugated to an antibody or portions of antibody (59). Similar chemistries to those used in nanoparticle conjugation have been utilized with liposomes, all aiming to attach an antibody or ligand to the outer shell of the liposome. Biotinylated phospholipids and avidin have been used in the liposomal formation, which can then be used to bind biotinylated antibodies. Covalent attachment of a thiolated antibody through a thioether bond formed with the maleimide group in maleimide-derivatized phospholipids has been described. Similar covalent attachments using carbodiimide between amine groups of the phospholipid or maleimidated phosphatidylethanolamine and carboxyl moieties of antibody have been studied (60).
Various studies have demonstrated the potential for antibody-conjugated colloidal systems, of both solid nanoparticle and liposomal formulation. These include the application of biotinylated CD3-specific antibodies––which recognize T-cell leukemia cells and primary T-lymphocytes––that were conjugated to thiolated gelatin nanoparticles through neutravidin-biotin noncovalent bonds. In vitro studies showed a selective uptake (by receptor mediated endocytosis) of these conjugated nanoparticles in 84% of a T-lymphocyte population. PEG-coated liposomes conjugated with folate on their outer surfaces were targeted to folate receptor–expressing tumors. In vitro results demonstrated enhanced cytotoxicity of encapsulated liposomal drugs through endocytotic liposomal drug uptake, whereas in vivo results have remained less encouraging (61). Wheat germ agglutinin has been conjugated to PLGA nanoparticles loaded with paclitaxel for localized pulmonary delivery. In vitro results showed that these nanoparticles exhibited enhanced cytotoxicity through improved cellular uptake through wheat germ agglutinin receptor-mediated endocytosis (62). Similarly, a Her2 receptor–specific antibody (trastuzumab) was conjugated to the surface of gelatin-human serum albumin nanoparticles through avidin-biotin noncovalent bonding. In vitro results showed effective internalization of the nanoparticles by Her2-overexpressing tumor cells via receptor-mediated endocytosis. A selectin ligand [Sialyl-Lewis (SLex)]- and ICAM-1–specific antibody recognizing endothelial-expressed inflammation markers were conjugated to PLGA microspheres and acted as leukocyte mimetics for targeting drugs to the vasculature in inflammatory diseases (63). OX26 MAbs were conjugated to the surface of liposomes that contained daunomycin and were used to bypass P-glycoprotein–mediated effects in multi-drug resistant RBE4 cells. Subsequent studies revealed brain accumulation of daunomycin from OX26 immunoliposomes to be higher as compared to brain accumulation of free (unconjugated) drug (64). Additionally, immunoliposomes containing doxorubicin conjugated with scFv fragment A5 (scFv A5) directed against human endoglin showed an increase cytotoxicity towards endothelial cells compared to non-targeted liposomes and free drug in vitro (65).
Clearly, these examples show there is a verifiably enhanced cellular response brought about by targeted colloidal delivery, albeit during in vitro experimentation. The challenge now remains for these findings to be applied successfully to clinical situations such as those demanding specific delivery of cytotoxic agents to multifocal sites of neoplastic growth.
Perspective
The advantages to be gained from the targeting of drug substances to specific cellular targets are apparent. However, the need to advance from drug-based therapies that rely primarily on pharmacodynamic and pharmacokinetic drivers to dictate drug distribution is pressing, certainly within the field of oncology. The difficulties encountered in current chemotherapy of neoplastic disease, such as the emergence of dose-limiting side effects, support this contention. Moving away from drug distribution dictated by physicochemical considerations (e.g., bioavailability and drug solubility) has been a difficult objective to achieve. Innovative strategies that exploit the innate ability of antibodies to attach to an antigen hold most promize in achieving this. The challenge remains that an antibody-conjugate assembled in such a way to transport a therapeutic payload to a cellular site must meet demanding criteria, including specificity, safety, and stability. Already, conjugates based on protein-antibody and antibody-colloid constructs have been described. The full translation of these technologies to the clinical environment remains the next big challenge.
Acknowledgments
We would like to thank Roberta Burden’s critical reading of this review. Work in the laboratory of CS and BW is supported by the BBSRC and The Royal Society.
- © American Society for Pharmacology and Experimental Theraputics 2005
References

Christopher Scott, PhD, (right, bottom) is also a lecturer within Pharmacy, QUB. His research focuses on the development of therapeutic antibodies and antibody-related strategies for the targeting and inhibition of tumor proliferation. To whom all correspondence should be addressed at E-mail: c.scott{at}qub.ac.uk; fax: +44 (0) 28 90247794.

Brian Walker, PhD, (right, middle) is Professor of Biomolecular Sciences, School of Pharmacy, QUB, working in collaboration with CS on novel applications of antibody technology for disease treatment.

Shane Olwill, PhD, (left, bottom) and Richard Buick, BSc, (right, top) Research Director and Senior Scientist, respectively, in Fusion Antibodies Ltd., a biotechnology company dedicated to the production of antibodies for use in a range of research, diagnostic and therapeutic applications.

Paul McCarron, PhD, (left, top) is a lecturer within the School of Pharmacy, Queen’s University of Belfast, Northern Ireland. His research focus is on the development of therapeutic nanoparticles, and Waleed Marouf, BSc, (left, middle) is currently undertaking his PhD under Paul’s supervision.

