Pin-pointing APP Processing
The neuropathological hallmarks of Alzheimer Disease (AD) are neurofibrillar tangles composed of tau protein and neuritic plaques containing the amyloid β-peptide (Aβ) that is derived from proteolytic processing of the amyloid precursor protein (APP). Either α- or β-secretases initiate the breakdown of APP by cleaving within its luminal domain, generating the soluble N-terminal fragments sAPPα or sAPPβ, respectively, as well as membrane-anchored C-terminal fragments (CTFs). Subsequently, CTFs are cleaved by the γ-secretase complex within the transmembrane domain, resulting in the production of either p3 (from αCTF) or Aβ (from βCTF) (1–3). β-Amyloid protein converting enzyme 1 (BACE1) has been identified as a membrane-bound aspartyl protease that fulfills all known criteria for a β-secretase. Inactivation of the BACE1 gene in mice abrogates the formation of Aβ and amyloid deposits, suggesting this enzyme as therapeutic target in treatment of AD (4, 5). So far, it has not been possible to provide solid evidence for a genetic linkage between the BACE1 gene and AD, but both the expression and activity of BACE1 protein are significantly increased in brain regions most affected in AD, supporting its role in neurodegenerative processes (6).
In cells, BACE1 is primarily located within the Golgi and endosomal compartments where the acidic milieu is optimal for its proteolytic activity. How APP is delivered to these acidic compartments for amyloidogenic processing remains controversial. Now, new findings on the role of the prolyl isomerase Pin1 in APP metabolism suggest an intriguing mechanism as to how this enzyme may influence delivery of the precursor protein to BACE1. Pin1, originally identified as a protein implicated in cell cycle regulation (7), is a member of a class of peptidylprolyl cis/trans isomerases (PPIases) that catalyze rotation of the protein backbone at proline residues. Subsequent studies revealed that Pin1 binds to and isomerizes distinct proline residues located in the commonly found phospho-Ser/Thr-Pro motif in a subset of proteins (8). Pin1-mediated isomerization affects target protein function and presents a regulatory mechanism for controlling many physiological processes such as cell proliferation and differentiation [reviewed in (9)]. Interestingly, the expression of Pin1 is decreased in degenerating neurons of AD patients and causes age-dependent neurodegeneration when deleted in mice (10, 11).
Phosphorylation of proteins on serine or threonine residues that precede proline is a key control mechanism for many biological processes. Proline is the sole α-imino acid in nature’s repertoire and, therefore, the only peptide residue that can exist in cis or trans conformation. When phosphorylated on serine or threonine found adjacent to proline, both tau and APP undergo conformational changes that promote the formation of tangles and, in the case of APP, processing to Aβ (12). The underlying mechanism was recently investigated by Pastorino and colleagues (12) who reported that Pin1 binds to full-length APP when APP is phosphorylated on Thr668 [residue number according to amino-acid (aa) sequence identified in the 695 aa APP variant (APP695)] and that Pin1 activity favors the production of the trans-conformation of Pro669. They also showed that in cultured cells Pin1 colocalizes and coimmunoprecipitates with APP and that overexpression of Pin1 reduces Aβ production. Taken together with other observations that genetic deletion of Pin1 in cells decreases sAPPα secretion, these findings suggest that Pin1-activity shifts cellular selectivity toward the non-amyloidogenic processing of APP (12). These data contrast to previous work that suggested an inverse relation existed between Pin1 levels and Aβ production (13), perhaps owing to the use of a different cell culture system and warranting further clarification.
To further demonstrate the physiological relevance of Pin1 in APP metabolism, Pastorino et al. also investigated amyloid production in both young and old Pin1-deficient mice. Although no difference was observed between 2–6 months of age, fifteen-month-old Pin1-null mice showed a ~30% increase in insoluble Aβ42, an APP proteolytic cleavage product closely associated with the development of AD. These findings were verified by breeding Pin1-null and Tg2576 mice [a mouse model of AD that expresses the Swedish mutation of APP (APPK670N, M671L) in large amounts under control of the hamster prion protein promoter]. Even as early as six months of age, offspring mice (Pin1–/–/Tg2576) produced ~50% more of the insoluble Aβ42 than did age-matched control littermates, providing further evidence for an in vivo role of Pin1 in AD (12). The findings are even more intriguing in light of recently published polymorphisms in the Pin1 promoter that result in low protein expression and links with onset of AD (14).
The question remains: How does isomerization of Pro669 in the cytoplasmic region of APP affect processing by α- and/or β-secretases such that the bulk production of cleavage peptides shifts from those associated with amyloidogenic pathways to those more favorably associated with non-amyloidogenic pathways? One possible explanation might lie in a differential effect that isomerization has on the APP cytoplasmic tail’s ability to associate with cytoplasmic binding partners that control trafficking and intracellular localization of APP (15). Binding of such adaptors is strongly dependent on transient ordering of the APP tail (16) (including rotation at residue Pro669) that, indeed, can be induced by the specific phosphorylation of Thr668 (17). One such candidate adaptor is Fe65, a neuronal protein that binds to the cytoplasmic tail of APP in a phosphoThr668-dependent manner (18). Fe65 is believed to enhance amyloidogenic processing (19) by linking APP to low-density lipoprotein-receptor-related protein 1 (LRP1) (20), an endocytic receptor of the LDL receptor gene family that rapidly undergoes endocytosis (as compared to other lipoprotein receptors that exhibit significantly slower rates of endocytosis), moving bound APP molecules quickly from the cell surface into the cell (21), thus delivering APP to endosomal compartments for BACE1 (and subsequent γ-secretase) cleavage and Aβ generation (22). One may speculate that in healthy neurons, efficient association of Pin1 with phosphorylated APP promotes fast isomerization to a trans conformation that can be dephosphorylated to a less amyloidogenic form of APP (22). In the diseased brain, however, where one finds significantly elevated levels of the Thr668 phosphorylated APP (22), insufficient Pin1 expression may impair conversion to the trans isomer and dephosphorylation (23). Thus, Pin1 and Fe65 may represent competing pathways for the proteolytic processing of APP, where Pin1 interaction diverts APP away from β-secretase but Fe65 interaction promotes β-site cleavage as shown in Figure 1⇓. The schematic diagram, however, is a simplified working model only, because the in vivo regulation of APP processing is a complex matter further underscored by the findings that both Pin1 and Fe65 actions are cell type–specific (13, 18).
In conclusion, new findings discussed here suggest future research aimed at increasing Pin1 activity in the brain as a therapeutic approach in AD; however, caution should be taken because more than thirty-five Pin1-binding proteins have been identified (with more likely to be demonstrated), and not all enzyme actions may be as beneficial as isomerization of APP. On the other hand, this multi-faceted enzyme might prove a magic bullet for therapeutic purposes to target different steps in neurodegeneration, including its observed ability to restore the function of AD-associated tau protein (23).
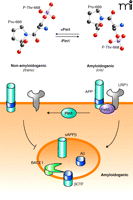
Pin1 promotes isomerization of the phosphorylated APP cytoplasmic domain and non-amyloidogenic processing. The delivery of APP to endosomes for β-amyloid protein converting enzyme 1 (BACE1)–mediated processing proceeds by low-density lipoprotein-receptor-related protein (LRP1)-mediated endocytosis. For this to occur, APP is likely indirectly linked to LRP1 through Fe65 (acting as an adaptor) binding to the tail of APP in a phosphorylation-dependent manner. Pin1 also binds to the tail of APP, especially to the phosphorylated Thr668-Pro669 in cis conformation, and catalyzes conversion to the trans isomer that undergoes rapid dephosphorylation, thus promoting the non-amyloidogenic processing of APP. Enhanced Pin1 activity could also prevent endocytic delivery of APP to BACE1 by competing with the action of Fe65, an activity that may present a novel target to decrease amyloid production.
- © American Society for Pharmacology and Experimental Theraputics 2006
References

Olav M. Andersen, PhD, is a research associate who has worked with Thomas Willnow at the MDC since 2001. That same year, he received his PhD in Molecular Biology from the University of Aarhus, Denmark, for his work on ligand binding determination by lipoprotein receptors. He is recipient of an American Health Assistance Foundation pilot award (2004) and is currently working on elucidating the molecular transport and processing mechanisms of APP. E-mail: o.andersen{at}mdc-berlin.de; fax: +49-30-9406-3382.

Thomas E. Willnow, PhD, is a graduate in Biology from the University in Munich, Germany, where he also obtained his PhD in Biochemistry in 1992. After a post-doctorate at the University of Texas Southwestern Medical Center in Dallas, he joined the Max-Delbrueck-Center for Molecular Medicine (MDC) in Berlin as junior faculty member. In 2000, he was appointed Full Professor for Molecular Cardiovascular Research. Presently, he is Professor for Molecular Cardiovascular Research at the Charité, Medical Faculty of the University of Berlin, and coordinator of the Cardiovascular Research Program of the MDC. E-mail: willnow{at}mdc-berlin.de

