Cannabinoids Biology: The Search for New Therapeutic Targets
Abstract
Cannabinoids, in the form of marijuana plant extracts, have been used for thousands of years for a wide variety of medical conditions, ranging from general malaise and mood disorders to more specific ailments, such as pain, nausea, and muscle spasms. The discovery of tetrahydrocannabinol, the active principal in marijuana, and the identification and cloning of two cannabinoid receptors (i.e., CB1 and CB2) has subsequently led to biomedical appreciation for a family of endocannabinoid lipid transmitters. The biosynthesis and catabolism of the endocannabinoids and growing knowledge of their broad physiological roles are providing insight into potentially novel therapeutic targets. Compounds directed at one or more of these targets may allow for cannabinoid-based therapeutics with limited side effects and abuse liability.

Introduction
Cannabis sativa, more commonly known as the marijuana plant, produces a complex mixture of medicinal substances that are collectively referred to as the cannabinoids. Despite the widespread use of marijuana and considerable research effort into its physiological actions, the biological mechanisms of the cannabinoids are just now beginning to be revealed with both expected and unexpected results. Marijuana’s most active component, tetrahydrocannabinol (Δ9-THC) (1), acts in animals as an agonist at the cannabinoid family of G protein–coupled receptors (GPCRs) that currently contains two subtypes, CB1 and CB2 (Table 1⇓). Following the cloning of the receptors for Δ9-THC (2–4), an endogenous lipid agonist for both the CB1 and CB2 receptors was discovered in mammals and named “anandamide,” from the Sanskrit word ananda for inner bliss and tranquility (5). Additional endocannabinoids continue to be discovered that are also cannabimimetic in central and peripheral tissues (6–9).
Biological Characteristics of the CB1 and CB2 Receptors
Unlike monoamine neurotransmitters that undergo vesicular release at synapses, endocannabinoids are lipid in nature and are released by hydrolytic enzymes from membrane phospholipid precursors (10–16). Subsequent to endocannabinoid release, signal termination appears to require a specific reuptake protein, or transporter (17–22), that works in concert with fatty acid amide hydrolase (23), monoacylglycerol lipase (24), or other uncharacterized enzymes that hydrolyze endocannabinoids. The various proteins involved in endocannabinoid release, physiologic action, and disposal offer intriguing opportunities for targeted drug development. Considering that marijuana has been used for medicinal purposes for thousands of years, only a single selective therapeutic compound, designed to block the CB1 receptor, has reached final stages of regulatory approval (25–29). This review will provide an overview of our current understanding of cannabinoid biology with a particular focus on the anandamide reuptake mechanism that has received much recent attention.
Cannabinoid Pharmacology
Marijuana has been used for thousands of years for both medicinal and recreational purposes, and yet only relatively recently have we begun to understand the basic mechanisms of its action in the brain and periphery. The phytocannabinoids derived from marijuana consist of over fifty potentially bioactive compounds; however, early cannabinoid research tended to focus on the principle bioactive component of marijuana, Δ9-THC (1). Once ingested, Δ9-THC generates seven major metabolites and approximately twenty-five potentially bioactive metabolites. The complexity of the compounds to which patients are exposed, in combination with the inconsistent dosing regiments inherent in smoking or ingestion, has made the interpretation of clinical trials with medical marijuana particularly challenging. Notwithstanding these challenges, there is little argument that exposure to Δ9-THC can offer medical benefits including anti-nausea and -emesis (30), appetite stimulation (31), analgesia (32), anxiolytic activity (33), anti-spasmodic activity (34), and lowering of intraocular pressure in glaucoma (35). However, psychotropic and addiction-related side effects have restricted its medicinal use. Less well-known but significant side effects of marijuana use include sedation, cognitive dysfunction, tachycardia, postural hypotension, dry mouth, ataxia, reduced fertility, and immunosuppression(36).
Until the last decade, the design of Δ9-THC-mimetic drugs concentrated on the development of triterpenoid cannabinoid agonists with appropriate metabolic stability and oral bioavailability but without significant side effects. In the early 1970s, Pfizer (New York, NY, USA) synthesized levonantrodol, a compound more potent than Δ9-THC, for the treatment of emesis associated with chemotherapy and postoperative pain. The development of levonantrodol was abandoned, however, owing to psychotropic side effects (e.g., dysphoria, dizziness, thought disturbance, and somnolence) that appeared coincident with efficacious doses. Eli Lilly and Company (Indianapolis, IN, USA) similarly produced the Δ9-THC analog, nabilone, which proved effective against nausea and vomiting in chemotherapy as well as anesthesia after abdominal surgery and radiation therapy yet was similarly dysphoric (37–39). Although nabilone (Cesamet) has been used successfully in the UK and Canada for over twenty years with no significant drug abuse problems (40), it remains scheduled as a narcotic by the Food and Drug Administration. Currently nabilone, marinol (a synthetic Δ9-THC marketed by Unimed Pharmaceuticals, Buffalo, IL), and Sativex (marijuana plant extract spray marketed by GW Pharmaceuticals, London, UK) are the only approved cannabinoid-based medicines.
More recently, Δ9-THC analogs such as the tricyclic benzopyran HU 210 [(6aR,10aR)-3-(1,1-dimethylbutyl)-6a,7,10,10a-tetrahy-dro-6,6-dimethyl-6H-dibenzo[b,d]pyran-9-methanol], the bicyclic CP-55,940 [5-(1,1-dimethylheptyl)-2-(5-hydroxy-2-(3-hydroxypropyl )cyclohexyl)phenol], and the amino-alkylindole WIN 55,212-2 [(R)(+)-[2,3-dihydro-5-methyl-3[(4-morpholinyl)methyl]pyrrolo[1,2,3-de]-1,4-benzoxazinyl]-(1-naphthalenyl) methanone mesylate salt], have been characterized as highly potent cannabinoid receptor agonists. The CB1 and CB2 receptors show comparable affinity for many cannabinoid agonists, including Δ9-THC, CP-55,940, HU 210, WIN 55,212-2, levonantradol, and nabilone; however, selective agonists and antagonists have been synthesized for both receptors (Table 2⇓), providing useful pharmacological tools that supplement the avail-ability of CB1, CB2, and CB1/CB2 knockout mice (41–45). These tools will be essential in elucidating the pharmacology and physiology of cannabinoid action and in engineering therapeutic molecules with minimal side effects.
Ligands Identified for the CB1 and CB2 Receptors
Cannabinoid Receptors
Subsequent to the discovery of a Δ9-THC binding site in rat brain tissue (2), two major receptor subtypes (CB1 and CB2) emerged from molecular genetics experiments (3, 4). A splice variant of the CB1 receptor (CB1a) has also been described, which appears to be expressed at low levels in rodents but not expressed in humans (46). A third putative cannabinoid receptor, GPR55, which binds cannabinoid ligands selectively, has recently been described (47); however, additional characterization will be required to identify GPR55 within the cannabinoid receptor family as CB3. The broad expression profile of cannabinoid receptors in the central nervous system (CNS) and periphery, along with the clinical data derived from studies with Δ-9-THC and CB1 antagonists, suggests that the targeting of specific cannabinoid receptors or their downstream signaling pathways will be an essential consideration in drug development (Table 1⇑).
The amino acid sequence of the CB1 receptor is relatively conserved across several species, including mammals, fish, hydra, mollusk, leech, and sea urchin (48–52), but is not thought to be expressed in insects (53). CB1 receptors are found predominantly in the presynaptic terminals of the CNS; however, they are also found in lower abundance in the periphery, including the immune system, testis, vascular endothelium, small intestine, liver, and peripheral nerve synapses (54–57). Although CB1 receptors are found throughout the brain, they are most dense in the cortex, hippocampus, basal ganglia, cerebellum, and spinal cord; this distribution is consistent with the effects of cannabinoids on memory, cognition, movement, and nociception (58, 59).
In contrast to CB1, the CB2 receptor is primarily expressed on immune cells in the periphery and acts to modulate immune function (4, 60). CB2 receptors are also expressed in tonsils, bone marrow, thymus, pancreas, adult rat retina, and peripheral nerve terminals in the mouse vas deferens (61). Particularly high expression levels are found on B cells and natural killer cells. CB2 signaling is thought to mediate cannabinoid inhibition of T cell proliferation, modulation of proinflammatory cytokine secretion, and B cell responses (62). Although originally thought to be absent in the CNS (4), CB2 receptor mRNA has been detected in cerebellar granule cells (63) and is upregulated in nervous tissue following inflammatory activation (64, 65). Recent evidence, in fact, shows significant CB2 receptor expression in the CNS, particularly in regions of the brain stem (66). Many ligands (especially those described earlier in the literature; see Tables 1⇑ and 2⇑) do not distinguish between the CB1 and CB2 receptors, despite only 44% overall amino acid sequence identity. This commonality of ligand binding, however, may be explained by the similarity (i.e., 68% identity) of their orthosteric ligand binding domains (4). In any case, selective CB2 ligands have become an area of active investigation (Table 2⇑).
At the molecular level, both CB1 and CB2 receptors predominantly signal through activation of Gαi/o proteins thereby resulting in the inhibition of adenylyl cyclase and reducing cAMP levels (67, 68). CB1 receptor activation is also linked to inhibition of N- and Q-type Ca2+ channels; activation of mitogen-activated protein kinases (MAPKs); expression of immediate early genes (e.g., krox-24); and activation of inwardly rectifying K+ channels. CB2 receptors display signaling similar to CB1 receptors, including MAPK activation; however, in contrast to CB1 receptors, CB2 receptor activation has not been shown to affect ion channel function (Table 1⇑) (62, 68).
Endocannabinoids
The discovery that the phytocannabinoid Δ9-THC binds to specific receptor proteins to elicit intracellular signaling events was an early indicator of the existence of one or more endogenous compounds that could exert neurotransmitter or hormonal control over central and peripheral CB1 and CB2 receptors. Endocannabinoids are now recognized as significant intracellular lipid signaling molecules that act in the central and peripheral nervous systems to regulate physiological, behavioral, and emotional functions. The first endocannabinoid identified, from porcine brain tissue, was anandamide (N-ara-chidonoylethanolamide; i.e., the fatty acid arachidonic acid coupled through an amide bond to ethanolamine) (5). Anandamide was subsequently isolated from human brain (69) and has been shown to mimic cannabinoid agonist activity in vitro (70, 71) and in vivo, capable of eliciting the classical tetrad of Δ9-THC-induced effects (i.e., analgesia, catalepsy, hypothermia, and hypomotility) (72).
Since the discovery of anandamide, a number of fatty acid–containing molecules with full or partial agonist activity at CB1 and/or CB2 have been either extracted from native tissues or chemically synthesized. These molecules, however, do not selectively modulate cannabinoid receptor signaling; they affect a variety of proteins, including ion channels (e.g., TRPV1), GPCRs (e.g., serotonin receptors), and enzymes (73, 74). The derivation of lipid messenger molecules from long-chain polyunsaturated fatty acids is reminiscent of the eicosanoid and prostanoid families of bioactive molecules. It is likely that many more lipid mediators will be discovered within the lipidome.
Besides anandamide, the most widely studied endocannabinoids are 2-arachidonoyl glycerol (2-AG) (6), 2-arachidonyl glyceryl ether (noladin ether) (8), and virodhamine (9). Levels of 2-AG in the brain are frequently one or two orders of magnitude higher than anandamide levels, although the physiological relevance of this concentration difference is unclear. Future research is needed to define cannabinoid receptor–specific neurotransmitters in terms of receptor type selectivity, localization of proteins involved in transmitter synthesis and degradation, relevant local concentrations proximal to receptors, and signaling crosstalk with other neurotransmitter systems.
Unlike neurotransmitter molecules that are typically held in vesicles prior to synaptic release, anandamide is synthesized on demand within the plasma membrane. One of the prevailing pathways for synthesis and release of anandamide begins with the transfer of arachidonic acid from the sn-1 position of rare phospholipids to the sn-3 position of phosphatidylethanolamine, thereby creating N-arachidonoyl phosphatidylethanolamine (NAPE). The reaction is catalyzed by a Ca2+-dependent N-acyl transferase (NAT) activity. A specific Ca2+-dependent enzyme then hydrolyzes NAPE’s phosphodiester bond (11, 14, 16, 75), releasing anandamide into the synapse from either the pre- or postsynaptic plasma membranes (Figure 1⇓) (76–78). Although originally called NAPE-PLD, this enzyme is a member of the zinc metallohydrolase family of enzymes. Alternative synthetic pathways have been hypothesized for anandamide, including sequential release of arachidonic acid and ethanolamine from phosptidylethanolamine through the action of phospholipase A2 and lysophospholipase D, respectively, and then condensation to anandamide through the action of a synthase-like enzyme (79). It is possible that different tissue- or cell-specific synthetic routes exist for fatty acid amide or glycerol ester endocannabinoids.
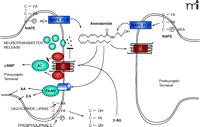
The Endocannabinoid Synapse NAPE (N-arachidonoylphosphatidyl ethanolamine) is hydrolyzed by NAPE phospholipase D (NAPE-PLD) to release free anandamide (AEA). Alternatively, precursor phospholipids are hydrolyzed by diglyceride lipase and phospholipase C to release the resident fatty acid (FA) from the sn-1 position and phosphatidyl-ethanolamine (p-EA) from the sn-3 position, respectively, yielding 2-arachidonoylglycerol (2-AG). Anandamide and 2-AG are agonists at the CB1 receptor. Anandamide can also be released from postsynaptic terminals to signal in a retrograde fashion to presynaptic CB1 receptors. The CB1 receptor either blocks or stimulates adenylate cyclase (AC) depending on cell type. CB1 receptors block neurotransmitter release from the presynaptic terminal. Disposal of anandamide, to yield EA and arachadonic acid (AA), and 2-AG is via movement across the plasma membrane followed by hydrolysis by cytoplasmic fatty acid amide hydrolase (FAAH). As discussed in the text, this movement of anandamide may involve a protein transporter (blue cylinder) that may work in conjunction with FAAH. 2-AG is also transported across the plasma membrane by a similar process and hydrolyzed by monoacylglyceride lipase.
Fatty acid esters such as 2-acylglycerol are generated from phosphatidylinositol precursors and removal of sn-1 and sn-3 groups from the glycerol backbone. Two major synthetic pathways for such fatty acid esters have been proposed. The first pathway involves removal of the inositol head group via phospholipase C and subsequent deacylation with sn-1 diacylglycerol lipase. The second pathway utilizes phospholipases A1 and C to remove sn-1 and sn-3 constituents. Whereas phospholipase C has been known for some time, sn-1 diacylglycerol lipases that are relevant to 2-AG formation have only recently been cloned and characterized [(12); for review, see (80)].
Anandamide Uptake and Hydrolysis
Termination of anandamide signaling appears to involve a two-step process beginning with transport across the plasma membrane followed by enzymatic hydrolysis into arachidonic acid and ethanolamine by cytoplasmic FAAH (10, 17, 19, 23, 81, 82). Functional anandamide transport activity has been well characterized in several cell types derived from rat, mouse, and human tissues (Table 3⇓). Most cells display a rapid (t1/2 = 2.5 – 4 min) (21, 83), saturable (Km = 0.190 – 45 μM), temperature dependent (Q10 = 1.6) (83), and enantioselective (18) mechanism for transport of anandamide; only a minor component of anandamide uptake is nonsaturable and occurs by diffusion. Reversible transport has been demonstrated in cells preloaded with radiolabeled anandamide (17). Evidence from structure–activity studies using anandamide analogs indicates that the transport process in whole cells has narrow structural requirements (18, 84–86), supporting the hypothesis that anandamide uptake occurs via a protein carrier–mediated process. However, pharmacological characterization of the transport process has been hampered by the paucity of selective inhibitors. Inhibitors that are analogs of anandamide, such as AM404 (IC50 = 1 μM) (19), inhibit both anandamide uptake and FAAH activity, suggesting that FAAH and the transport protein share a structurally similar binding site (87–89). Several groups have reported substances that selectively inhibit the cellular uptake of anandamide with no apparent effect on FAAH activity (87–92), but it is not clear whether these compounds truly represent selective inhibitors or merely reflect limited access to cytoplasmic FAAH. A recent publication attempted to provide an assessment of the ability of putative inhibitors to block functional anandamide uptake, FAAH enzyme activity, and transport protein binding and concluded that no selective tools currently exist for either FAAH or transport inhibition (93). Similar conclusions were made by other investigators with a selected group of putative selective inhibitors (94).
Summary of Km and Vmax of Anandamide Transport
Molecular Models of Anandamide Uptake
Several hypotheses of transport of anandamide-like endocannabinoids have evolved, with varying levels of validation (95, 96). The most direct hypothesis involves the passive diffusion of lipophilic endocannabinoids across the plasma membrane. Alternatively, an endocytotic uptake mechanism has been proposed, based on the cytoplasmic and predominantly perinuclear localization of FAAH. In addition, plasma membrane–localized FAAH alone may be all that is required to gather and dispose of extracellular anandamide (23). Using a novel small-molecule inhibitor of anandamide uptake, our own group has identified a high-affinity binding site that is independent of FAAH expression, indicating a role for a specific protein-mediated uptake process (22). Hypotheses for anandamide uptake generally fall into one of four models described in greater detail below. Although some evidence exists supporting the transport of other endocannabinoids through the same or a similar protein as that for anandamide (97–99), less is currently known about transport and metabolism of other endocannabinoids, so they will not be discussed here.
Model 1: Plasma Membrane–Associated FAAH
FAAH appears primarily localized within perinuclear membranes and is not found at the plasma membrane. Immunohistochemical studies of the rat CNS reveal punctate cytoplasmic inclusions of FAAH within the cell bodies and dendrites of pyramidal and Purkinje cells (100, 101), and confocal fluorescence microscopy identifies punctate FAAH immunoreactivity in perinuclear areas (102). More recently, the physical separation of FAAH from the plasma membrane has been taken to suggest that transport is independent of hydrolysis (82). However, these studies cannot rule out an undetected but physiologically relevant level of FAAH at the plasma membrane more proximal, perhaps even adjacent, to the transport protein that would allow for concerted anandamide uptake and hydrolysis (Figure 2⇓).
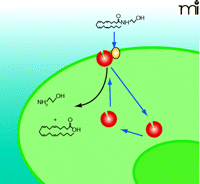
Anandamide Transport by Plasma Membrane-Associated FAAH (Model 1). This model of transport requires fatty acid amide hydrolase (FAAH, red) alone to bind anandamide from the extracellular space directly. The curved black arrow represents FAAH-mediated hydrolysis of anandamide into ethanolamine and arachidonic acid. See text for details.
Hydrolysis of anandamide by FAAH appears to be the primary driving force for its cellular uptake. Brain extracts from FAAH-deficient mice display 100-fold less anandamide hydrolysis activity than wild-type mice (103). Although FAAH preferentially hydrolyzes long chain fatty acid amides, such as anandamide and oleoylamide (23, 104–106), it also hydrolyzes 2-AG, an endocannabinoid structurally and pharmacologically similar to anandamide (107). However, due to the wide variation in reported Km values [0.8–180 μM (20, 104, 108–113) (Table 3⇑)], it is not clear if FAAH is a physiologically relevant hydrolysis pathway for 2-AG. FAAH knockout mice, for example, retain their ability to catabolize 2-AG (114), most likely through hydrolysis by monoacylglycerol lipase (24, 115).
The crystal structure of FAAH supports the presence of several domains that grant the enzyme access to both the inner leaflet of the plasma membrane and the cytoplasmic milieu (23, 116). The substrate channel is amphipathic, which may allow the admission and movement of the polar head group of anandamide into the active site of FAAH. Another channel is comprised almost entirely of hydrophobic residues thought to be responsible for substrate binding and recognition. A third channel creates a solvent-exposed cytosolic port (116). Following anandamide hydrolysis, liberated fatty acid and amine products can be envisaged to exit the enzyme via the membrane- and cytosolic-access channels, respectively (116). These structural features are consistent with a plasma membrane–associated enzyme that accepts anandamide directly from the extracellular space or through diffusional processes (see below), or possibly through protein-mediated facilitated diffusion as described below. These features are also consistent with perinuclear-localized FAAH, which would receive anandamide through passive diffusion or endocytotic transport (see below).
Model 2: Passive Diffusion Driven by FAAH
It has been proposed that anandamide accumulation is solely dependent on passive diffusion across the plasma membrane and that FAAH-mediated enzymatic cleavage of anandamide maintains the inward concentration gradient and drives continued anandamide uptake (102) (Figure 3⇓). In this model, lipophilic anandamide diffuses through the plasma membrane where it eventually integrates into FAAH-enriched perinuclear membranes (82). In concordance with this hypothesis, a specific transport protein has been elusive, and anandamide uptake is not readily inhibited by traditional uptake inhibitors as measured within five to thirty seconds (96, 102). However, measurements of anandamide uptake at short time points may reflect non-specific integration of highly lipophilic anandamide into the cell membrane rather than specific transport. In a recent review, the authors allowed for the possibility that diffusion accounts for the initial non-specific process, which may be followed by a specific uptake process (96). In addition, the recent identification of a binding site involved in anandamide transport has challenged the passive diffusion theory (22); however, a passive diffusion process may be relevant for signaling-independent absorption and recycling of lipid molecules in specific cell types. For example, FAAH-deficient cell types do exist and yet accumulate anandamide, albeit at significantly reduced rates compared to FAAH-expressing cells (117).
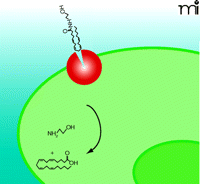
Passive Diffusion of Anandamide Driven by FAAH (Model 2). In this model, the sole function of FAAH (red) is to establish a concentration gradient of anandamide transport (blue arrow) through hydrolysis of anandamide into arachidonic acid and ethanolamine. FAAH is localized exclusively in the perinuclear area. Delivery of anandamide to perinuclear FAAH may require a special protein or process currently undefined.
Model 3: Endocytosis-Mediated Anandamide Uptake
Due to the lipophilic nature of anandamide and related endocannabinoids, it has been hypothesized that these molecules enter the cell via an endocytotic process rather than by diffusing through the plasma membrane and cytosol to reach the perinuclear FAAH (117–119) (Figure 4⇓). Conceptually, anandamide is sequestered in caveolin-rich lipid rafts in the plasma membrane and subsequently delivered to perinuclear FAAH. Lipid rafts are membrane micro domains that are enriched in cholesterol, sphingolipids, arachidonic acid, and plasmenylethanolamine (120, 121), and include a family of integral membrane caveolin proteins that serve as the major structural components for caveolae (122). Caveolae-related endocytosis is distinct from clathrin-dependent endocytosis that involves the internalization of specialized membrane domains known as clathrin-coated pits (123). A caveolae-related endocytotic mechanism for the cellular uptake of anandamide meets the criteria of being temperature-dependent, rapid, saturable, and energy-independent, and could also be facilitated by, but not dependent upon, FAAH activity (95). It is conceivable that specific plasma membrane–associated binding proteins may be enriched in caveolae, thereby facilitating the association of endocannabinoids with the endocytotic process. Endocytotic vesicular accumulation of anandamide is also compatible with the suggestion that inactivation could be mediated in part through accumulation of anandamide within a cytoplasmic compartment (124).
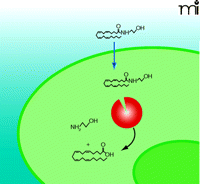
Endocytosis-Mediated Anandamide Uptake (Model 3). In this model, anandamide enters the cell through the plasma membrane via a rapid endocytotic process (short blue arrows), which may involve ancillary binding proteins (orange), and is delivered to cytoplasmic FAAH (red) for hydrolysis.
Model 4: Facilitative Diffusion Driven by FAAH
Similar to passive diffusion, facilitative diffusion of anandamide requires a FAAH-induced concentration gradient (Figure 5⇓). In facilitative diffusion, however, a protein transporter or carrier molecule is envisaged to move anandamide across the plasma membrane and into the cytoplasm. A recent study physically separated anandamide transport and hydrolysis activities via cell fractionation, suggesting a distinct FAAH-independent transport process (82). In addition, a radioactive derivative (i.e., 125I-LY2318912) of the potent anandamide uptake inhibitor LY2183240 was used in cells devoid of FAAH to reveal binding constants identical to those that characterize FAAH-expressing cells; these results are consistent with the presence of a specific transport-facilitating protein (22). Furthermore, bidirectional transport of anandamide has been demonstrated, suggesting that the putative endocannabinoid transporter may play a role in release as well as inactivation (17). Additional studies will be required to clone and characterize the molecular nature of the binding site.
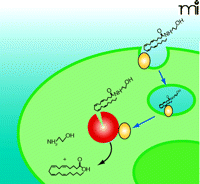
Facilitative Diffusion of Anandamide (Model 4). This model involves a specific plasma membrane binding protein (orange) that facilitates anandamide transport across the plasma membrane. Although the highest concentration of FAAH (red) has been localized to perinuclear compartments, our working hypothesis suggests that sufficient FAAH resides at the plasma membrane closely associated with the transport protein or moves to the plasma membrane based on use-dependency to facilitate transport and hydrolysis.
The endocannabinoid transport process appears to be different from better-characterized molecular transport processes. High-affinity plasma membrane transporters have been classified into predominantly two families based on topology, ion dependence, and sequence relatedness: the Na+- and Cl−-dependent transporter family and the glutamate transporter family. The ion-dependent family includes subfamilies for monoamine transporters [e.g., the dopamine transporter (DAT), norepinephrine transporter (NET), and serotonin transporter (SERT)] and for amino acid transporters [e.g., GABA transporters (GAT1–4), glycine transporters (GlyT1–2), proline transporter, and taurine transporter], whereas the glutamate transporter family consists of five isoforms (i.e., GLAST, GLT-1, EAAC1, and EAAT4–5) (125, 126). In contrast to other classic membrane bound transporters, the anandamide transporter appears to require neither an ion gradient nor ATP (17), and no cofactor has been identified as necessary for its function. The anandamide transport process is insensitive to inhibitors (e.g., bromocresol green, cocaine, and verapamil) of established transporters (21). Several members of the fatty acid–transport and –binding protein families have proven incapable of increasing anandamide uptake in cells (unpublished results, E. Barker, Purdue), which adds to the consensus that the anandamide transporter is a unique protein having little or no homology to classic transporter families.
Anandamide at the Plasma Membrane
Recently, translocases have begun to emerge as candidates for the anandamide transporter. Translocases are membrane bound proteins that “flip” aliphatic compounds from one leaflet of the membrane to the other, in a concentration-dependent manner; however, in contrast to the anandamide transporter (17), translocases are generally ATP-dependent. The anandamide transporter may be related to the family of translocases that includes the CD36/FAT protein that translocates arachidonic acid, a major structural component of the anandamide molecule (127).
Due to its amphipathic nature, the conformation of anandamide is an important determinant in its interaction with plasma membrane proteins. A recent study demonstrates that, once inserted into the plasma membrane’s outer leaflet, anandamide adopts an extended conformation, such that its polar ethanolamine head group lines up with the polar head group of the neighboring phospholipid within the membrane and the aliphatic tail points towards the bilayer center (128–130). The results of this study are consistent with the hypothesis that anandamide approaches the CB1 receptor binding site by fast lateral diffusion within the outer membrane leaflet, where it interacts with the hydrophobic groove formed by helices 3 and 6 of the CB1 receptor, thus activating the receptor (80, 131, 132). If it is indeed the case that anandamide laterally diffuses in the outer membrane leaflet to activate the CB1 receptor, then translocation of anandamide to the inner leaflet of the plasma membrane may be the step that serves, independent of hydrolysis per se, to extinguish its biological activity. A translocase similar to FAT/ CD36 would be a logical candidate to perform such a task.
Conclusion
The endocannabinoid transport process promises to be a useful therapeutic target to leverage cannabinoid receptor agonism with the potential to avoid psychtropic side effects. The question still remains as to how an anandamide transport protein and FAAH might interact in order to orchestrate the metabolism of anandamide. In a recent study, Oddi and coworkers (65) demonstrate that FAAH activity primarily resides in intracellular perinuclear membranes, whereas transport activity is localized almost exclusively in plasma membrane fractions. Immunostaining of live cells has demonstrated that FAAH is primarily localized in intracellular regions (133). The physical separation of these distinct activities compels the question: Does anandamide travel to the perinuclear membrane to meet FAAH, or does FAAH travel to the plasma membrane to meet anandamide? It is also not clear if anandamide diffuses through the cytoplasm, carried by a yet unknown binding protein, or if it is transported via endocytosis. Due to the lipophilic nature of anandamide, it is unlikely that it diffuses unaided through the cytoplasm to the perinuclear membranes, giving strength to an endocytic mechanism. It is also possible that trace levels of FAAH in the plasma membrane are sufficient to drive facilitative diffusion. Perhaps FAAH is stored in perinuclear membranes and translocated to the plasma membrane when stimulated by anandamide-mediated cannabinoid receptor signaling events.
The pending launch of the first bone fide cannabinoid therapeutic agent opens the door to the development of other possible therapeutic agents that may exploit cannabinoid mechanisms. Rimonabant is a CB1 receptor antagonist being developed for obesity and metabolic syndrome, promising to reduce appetite through dampening of the cravings for highly palliative foods and to positively modify lipid profiles. Cannabinoid receptors are abundant in both the CNS and periphery and fit into the well-heeled GPCR-centric approach to drug development. However, no group has yet achieved selective stimulation of either CB1 or CB2 receptors or developed a novel approach to providing cannabinoid receptor agonism while simultaneously avoiding psychotropic side effects. Additional targets are emerging including endocannabinoid reuptake blockade and concomitant augmentation of cannabinoid neurotransmission. Mechanistically, the result of reuptake or hydrolysis blockade should provide the therapeutic benefit of cannabinoid receptor activation in a more physiologically relevant temporal and spatial context. The hope is to avoid undesirable psychotropic and systemic side effects. Although the complexity of endocannabinoid signaling and mechanism of cannabinoid regulation have yet to be fully defined, they clearly warrant further exploration as means of therapeutically exploiting stimulation of the cannabinoid system while avoiding liabilities associated with the use of medicinal marijuana.
- © American Society for Pharmacology and Experimental Theraputics 2006
References

Steven Moore, PhD, received his doctoral degree from the Department of Cell and Structural Biology at the University of Illinois at Urbana-Champaign in 2003. He completed his postdoctoral training in the laboratory of Christian Felder in the Neuroscience Division of Eli Lilly and Company in 2005, and subsequently joined the R&D department of Pierce Biotechnology in Rockford, Illinois.

Amy K. Dickason-Chesterfield, MS, is an Assistant Senior Biologist in the Department of Neuroscience at Eli Lilly & Co. She is also a doctoral student at Miami University working under the guidance of Chris Felder and Phyllis Callahan. She attended Hanover College and Miami University for her undergraduate and graduate studies, respectively.

Christian Felder, PhD, completed his doctoral work in the Deptartment of Biochemistry at Georgetown University School of Medicine. He completed his postdoctoral training in the laboratory of Julius Axelrod at the NIMH in 1990. As Chief of the Unit on Cell and Molecular Signaling at the NIMH, he continued to work with Julius Axelrod investigating the pharmacology and signal transduction of G protein–coupled receptors with a focus on the muscarinic and cannabinoid receptor families. He is currently a Research Fellow in Neuroscience at Eli Lilly & Co., focusing on the neurobiology of psychiatric diseases.

