How Specific Are “Target-Specific” Drugs? Celecoxib as a Case in Point
Proteins can assume, theoretically, a near infinite number of shapes, and yet, the conformational space assumed by human proteins is relatively limited. Segments of proteins possessing favorable conformations are conserved in evolution and recur as domains in multiple genes encoding them. Moreover, the nature of physicochemical interactions––based on a limited number and diversity of amino acids––also leads to a finite, albeit large, number of possible structures, which is relevant to our understanding of how drugs and binding pockets on protein targets can interact.
Drug discovery is guided by attempts to produce the most selective and potent ligand for a “druggable target.” There are many examples of such “highly selective” drugs; however, given the number of proteins and potential drug-binding pockets encoded by the human genome, one might not be surprised if drugs touted as highly specific should turn out to interact with multiple targets.
Recently, a branch of the biotech and the pharmaceutical industry has begun to focus on the question of “how specific is ‘specific’?”, by performing a reevaluation of interactions between the most commonly used drugs and a large number of pharmaceutically relevant targets, such as receptors, enzymes, transporters, structural proteins, etc. To address these issues, a new discipline has emerged in the form of chemogenomics, i.e., the testing of large chemical libraries against multiple genomic targets (1–3). This approach has revealed unexpected drug-protein interactions, leading to the emergence of large databases containing multiple potential protein targets for drugs (4, 5). A better understanding of multiple parallel drug actions is imperative, particularly for widely used drugs already in clinical use. Following FDA approval, increasingly large numbers of patients exposed to the drug often reveal unexpected adverse effects that could lead to withdrawal from the market. Drug interactions with unsuspected protein targets represent one possible cause for the occurrence of such sporadic adverse effects, as shown for drug-induced arrhythmias caused by prolongation of Q-T intervals through binding to ion channels (6). In this case, routine screening of drug candidates is already being performed during drug development. Yet, drug safety and efficacy surveys (performed post-FDA approval) fail to account for discovery of newly revealed drug targets even where this has likely pharmacological and clinical implications.
The re-evaluation of celecoxib is a relevant example. Marketed as Celebrex®, this selective cyclooxygenase-2 (COX-2) inhibitor has potent anti-inflammatory properties and is widely used in the treatment of osteoarthritis and similar disorders. Following the discovery that the inducible COX-2 can function as a main mediator of inflammatory processes while the constitutive COX-1 has been associated with adverse effects such as gastrointestinal bleeding, the development of a new class of highly selective COX-2 inhibitors (Figure 1⇓) has become a milestone of modern drug discovery––and a multi-billion dollar market. Recently, however, and probably not surprisingly (given the rapidity of their wide-spread use), a serious side-effect has emerged in the form of increased risk for coronary artery events. Although this dangerous side-effect has been conclusively demonstrated thus far only for Vioxx® at relatively high doses over prolonged time periods, the fact that the assumed mechanism of adverse action also involves COX-2 activity has shed doubt over the entire class of COX-2 inhibitors.
There is much debate as to whether all COX-2 inhibitors are “equal” or whether there are substantive differences inherent in these inhibitors that affect their efficacy and risk for adverse effects. There may be several reasons that support the claim that several substantive differences exist. For example, small structural modifications–even very small ones–could alter the interaction of drugs with any of the numerous membrane transporters, and hence, might affect the relative distribution of drugs into their target tissues. Further, even the “highly selective” COX-2 inhibitors prove not to be so selective after all when one casts the net of drug-protein interactions more broadly.
At a recent conference, scientists from Iconix Pharmaceuticals (Mountain View, CA) divulged results obtained through massive screening of protein interactions with all top-selling drugs. We noted, to our surprise, that celecoxib was listed as a more potent inhibitor of carbonic anhydrase than of COX-2. Indeed, a brief scan of the scientific literature shows that both celecoxib and valdecoxib (Bextra®) have been known for some time to possess high affinity for the carbonic anhydrases as well as for COX-2 (7–9). Carbonic anhydrase inhibitors are in wide use for the treatment of glaucoma (aquamox) and high-altitude sickness, and may also reduce colon cancer risk. Although these findings represent beneficial actions under certain conditions, such inhibitors also affect electrolyte balance when taken chronically. Distinct human genes encode the fourteen known carbonic anhydrase isozymes, and we do not know how drugs distinguish between them; therefore, the pharmacological consequences of celecoxib acting as a carbonic anhydrase inhibitor are uncertain, yet highly probable. Nonetheless, Celebrex® appears to work well as an anti-inflammatory while possibly also reducing high-altitude sickness (uncontrolled personal case study by one of the authors). The merits of this dual action as COX-2 and carbonic anhydrase inhibitor, including any influence on coronary artery events, as well as potential consequences resulting from electrolyte imbalance, have not been reported in clinical studies and remain to be explored.
We think it important to address this putative dual action of celecoxib and of all COX-2 inhibitors, for that matter. It is not unreasonable to propose that if this dual action were to be substantiated under clinical conditions, an assessment of its clinical relevance is in order, given the wide-spread use of COX-2 inhibitors such as celecoxib. Moreover, a comparison between COX-2 inhibitors might reveal differences in relative potencies at the two enzymes that could also affect risk of adverse effects such as coronary artery disease. Although we single out celecoxib in this article, we wish to broaden the perspective of this discussion to propose that a much more systematic effort be devoted to unveiling drug-protein interactions, with the understanding that each drug has multiple protein partners. We already know the significance of these interactions in drug metabolism where the role of multiple cytochrome P450 (CYP) enzymes is studied in detail, as well as for drug transporters (although only a fraction of the countless transporters is currently considered). Insights into multiple drug targets could aid in prediction and prevention of side effects as well as provide direction for fine-tuning drug discovery and development where strategies should be based on knowledge of all potential mechanisms of action. Whether significant interactions are revealed during drug development or in post-marketing surveillance, the pharmaceutical industry, academia, and the FDA should act to understand the public health implications of such findings.
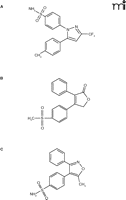
Chemical structures of “highly selective” cyclooxygenase-2 inhibitors. Celecoxib (A), rofecoxib (B), and valdecoxib (C) are shown.
- © American Society for Pharmacology and Experimental Theraputics 2006
References

Laura M. Bohn, PhD, is an Assistant Professor in the Department of Pharmacology, at The Ohio State University College of Medicine, and is a faculty member of the Integrated Biomedical Science Graduate Program and the Neuroscience Graduate Program. Her laboratory studies how regulation of the mu-opioid receptor, a member of the G protein–coupled receptor (GPCR) superfamily, determines the biological effects of opiate narcotics. E-mail: bohn.24{at}osu.edu; fax: 614-292-7232.

Wolfgang Sadée, Dr. rer. nat., is Felts Mercer Professor of Medicine and Pharmacology at The Ohio State University College of Medicine. He also serves as Director of the Program in Pharmacogenomics, and is a faculty member in Psychiatry, Medical Genetics, Pharmacy, OSU Comprehensive Cancer Center, and Davis Heart & Lung Research Institute. E-mail: wolfgang. sadee{at}osumc.edu; fax: (614) 292-7232.

