Future Directions in Cardiovascular Pharmacology: Examples from the Renin-Angiotensin System
Often, medicine is administered to cure a disease that affects one site of infection, one tissue, or organ system. There are, however, many instances where pathological changes in one system (i.e, one organ, or a set of organs connected by hormonal control, etc.) affect otherwise healthy systems that share similar regulatory mechanisms. For example, the cardiovascular system is interconnected with the regulatory system that governs blood glucose homeostasis. Although declining for several decades, the incidence of cardiovascular disease has recently begun to plateau. At the same time, the prevalence of type II diabetes and insulin resistance in the US has increased, giving reason to suspect that increased diagnoses of cardiovascular disease will rise concomitantly as the regulatory controls of the cardiovascular system are impinged on by the pathophysiology associated with diabetes and insulin resistance. Thus, future treatments should, if possible, be developed that have a positive outcome for multiple homeostatic mechanisms (1). The coexistence of disorders in multiple complex regulatory systems also argues for greater emphasis on a “systems and integrative pharmacology” approach in the renewed efforts of basic research to produce effective therapies.
Several cardiovascular disorders may be successfully treated through therapeutic interventions targeting the renin-angiotensin system (RAS). The RAS participates in inflammation, oxidative stress, signaling pathways interacting with metabolic hormones (such as insulin and leptin), body weight regulation, vascular growth and atherosclerosis, and coagulation and hematopoiesis. Thus, it is not surprising that blockade of the RAS may have widespread benefits. The issues facing advancements in RAS-targeted therapy, however, are similar to those pertinent to cardiovascular pharmacology in general. Many of these topics have been covered in recent issues of this journal, including approaches for tissue-specific administration of therapeutic agents (2, 3), use of enzymes or antibodies in the therapeutic regimens (4, 5) and gene therapy and other molecular approaches for treatments. Below are several key concepts that have been or should be considered in meeting the challenge for improving cardiovascular therapies in the near future, within the context of the pharmacology of the RAS.
The RAS is involved in the mechanisms underlying development of renal dysfunction, hypertension, heart failure, and atherosclerosis. Over the past decade, our understanding of the mechanisms of the RAS to promote inflammation and oxidative stress––and its interference with signaling pathways for metabolic hormones––has greatly improved, providing opportunities for clinical benefit independent of blood pressure–lowering actions that accompany the blockade of angiotensin (Ang) II. Indeed, the discovery of multiple receptor subtypes, active peptides, and new pathways for precursors of the active peptides has underscored the complexity of the RAS (Figure 1⇓). With regard to an activated RAS, we must now distinguish between prohypertensive Ang II and antihypertensive (or at least, non-hypertensive) Ang-(1-7). Ang-(1-7) acts at a distinct receptor to promote prostanoid synthesis and signaling and to activate nitric oxide systems and functions as a physiological antagonist of Ang II by opposing Ang II-mediated intracellular signaling pathways. In fact, recent studies indicate that aging, hypertension, fibrosis, and renal dysfunction may all stem from a deficit in Ang-(1-7), leading to pathology (6–12). Even with normal, basal amounts of Ang-(1-7), an increase of Ang-(1-7) during states of overexpressed (or overactive) Ang II provides significant cardiac and renal protection (13–16). This realization has led to re-evaluation of mechanisms underlying interruption of the RAS at different points in the cascade and has challenged the development of pharmaceutical agents or approaches that selectively shift the balance from Ang II to Ang-(1-7).
New targets and new approaches to old targets have been identified within the RAS. The use of renin inhibitors has recently resurfaced as a therapeutic approach to block or attenuate the RAS. Although effective in short-term studies to lower blood pressure (17), it remains unknown whether renin inhibitors will be associated with the many beneficial aspects of angiotensin converting enzyme (ACE) inhibition or Ang Type 1 receptor (AT1R) blockade. Prorenin, renin, and ACE may also act at distinct receptors to activate signaling pathways directly (18–20). The binding of ACE inhibitors reportedly activates signaling pathways that lead to nitric oxide formation. Prorenin binding to its receptor not only activates the enzyme making it capable of converting angiotensinogen to Ang I, but also activates mitogen-activated protein kinases (MAPKs) through a G protein–coupled mechanism. The physiological significance of these cellular events is an active area of investigation. Another newly identified component of the RAS is Ang-(1-12), a potential precursor to Ang II and Ang-(1-7) (21–23). Angiotensinogen is the only known candidate precursor for Ang-(1-12); Ang-(1-12) is not likely to arise from the actions of renin, nor does renin appear involved in further metabolism of Ang-(1-12) into smaller peptides (23, 24). Preliminary evidence does support further processing of Ang-(1-12) by ACE or neprilysin [also termed neutral endopeptidase (NEP)] (24). In the circulation and in isolated vasculature, Ang-(1-12) produces increases in pressure and vasoconstriction, respectively (i.e., actions that are blocked by AT1R antagonists or ACE inhibitors), suggesting conversion of Ang-(1-12) to Ang II (21). Each of the above-mentioned components might possibly emerge as a therapeutic target once their place in the pharmacology of the RAS is better defined.
The presence of angiotensinogen and renin are necessary for expression of Ang-(1-7). In fact, with either ACE inhibition or AT1R blockade the feedback inhibition of renin by Ang II is blocked and renal renin release is increased. Under these conditions, concentrations of Ang-(1-7) are elevated (25, 26). ACE2 is a recent addition to the angiotensin-processing enzyme family, converting Ang II to Ang-(1-7). ACE2 activity is elevated as a result of increases in mRNA and protein expression, with concomitant AT1R or ACE inhibition. In the presence of Ang II, ACE2 activity is decreased by events involving regulation of kinase and phosphatase pathways (27–31). Overexpression of ACE2 in tissues such as heart, brain, and vasculature has protective effects on the cardiovascular system (32–34). Increased circulating levels of Ang-(1-7) contribute as much as 30%–40% of the blood pressure lowering actions of AT1R or ACE inhibitor therapy (25, 26). Long-term administration of Ang-(1-7) also has protective effects independent of any blood pressure–lowering action, providing reno- or cardio-protection through reduction of inflammation and possibly oxidative stress (14–16). The anti-proliferative actions of the peptide are also under study as an adjunct for cancer chemotherapy and are accompanied by reduction of inflammation through decreases in cyclooxygenase-2 (COX-2) (35). What can be inferred from the above findings is that complete disruption of the RAS, such as through the action of a renin inhibitor, would not be associated with a healthy profile, unless the Ang-(1-12) pathway is available for formation of Ang-(1-7), or the loss of Ang II predominates.
All components of the RAS are present in a variety of extra-renal tissues and their regulation is independent of the circulating hormonal system. The predominant localization of prorenin or active renin and other enzymes required for subsequent processing of Ang I or Ang-(1-12) will dictate the final bioactive peptide products. Thus, the concept that local RAS blockade is essential to therapy has re-emerged as a factor in design of pharmaceuticals to enhance their effectiveness. In fact, preliminary studies reveal that antisera to Ang-(1-12) administered into the brain ventricles over a two-week period lowers blood pressure in a model of Ang II–dependent hypertension, an effect that may be additive with renin blockade (36).
Expression of the critical enzymes regulating the pathways leading to Ang II versus Ang-(1-7) also varies among tissues. Whether specific therapies intended to reduce Ang II expression, elevate Ang-(1-7) expression, or selectively block their receptors are able to access all tissues equally well, however, are unanswered questions. A recent study showed that the higher lipophilicity profile of certain RAS blockers was associated with better protection from age-related decline in cognitive function (37). Based on available lipophilicity data, one might assume that captopril, lisinopril, and ramapril would cross the blood-brain barrier more readily than enalapril, benazepril, or quinapril, for example. Agents that can be coupled with highly lipophilic molecules or that use selective transporters for entry into brain or other tissues would be of great interest. As described in a recent article (3), this is a difficult task given that the means of achieving transport or lipid solubility usually leads to a drug complex whose size impedes facile movement across the blood-brain or CSF-blood barriers.
Recently, several interesting studies have demonstrated the utility of gene therapy by overexpression of the antihypertensive components of the RAS and by knock-down of the prohypertensive components. Examples of the former include overexpression of Ang-(1-7) or ACE2 in heart or brainstem to lower arterial pressure in hypertensive animals (32, 34, 38, 39). Knock-down of AT1R or angiotensinogen expression by administration of antisense oligonucleotides or siRNA also leads to effective therapeutic control of arterial pressure in hypertensive rats (40–44). Of course, the local delivery of nucleotide constructs to the brain requires invasive procedures not conducive to patient treatments; thus, the above mentioned modifications would be needed to provide for tissue-specific administration or access.
The targeted disruption of the glial source of angiotensinogen by specific antisense oligonucleotides (45) has also been used to interrupt the brain RAS substrate, resulting in an interesting transgenic phenotype with enhanced sensitivity of the baro- and chemoreflexes, lower resting arterial pressure (45–47), indices of improved cardiovascular and metabolic function, a longer lifespan (47), and protection from cardiac damage in response to coronary artery ligation (48) and angiotensin II infusion–induced hypertension (49). Long-term systemic administration of AT1R blockers in rats (50) or either AT1 or ACE inhibition in humans (27, 51) mimics the phenotype of the transgenic ASrAogen (antisense RNA against angiotensinogen expression in astroglia) rats to a large extent. Thus, the contribution of inhibition of the brain RAS to the overall benefits with these therapies remains of interest (52).
Interference with the RAS provides improvements in multiple homeostatic systems, consistent with the need for interventions that control blood pressure as well as energy metabolism. Whether the Ang II blockade or the increase in Ang-(1-7) proves to be the more clinically beneficial for cardiovascular and metabolic diseases clinically has yet to be determined. Whether the other components, such as the renin receptor and Ang-(1-12), emerge as important additional targets for therapies awaits further investigation.
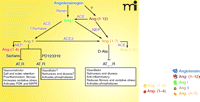
Schematic of the enzymes and processing of substrates for the renin-angiotensin system. Ang II is formed through the actions of angiotensin converting enzyme (ACE). Ang-(1-7) can be formed through several enzymatic reactions: 1) proteolytic cleavage of Ang I or Ang-(1-12)––a proteolytic cleavage product of angiotensinogen––by neprilysin, or 2) proteolytic cleavage of Ang II by ACE2. Note that neprilysin and ACE2 inactivate Ang II, whereas ACE inactivates Ang-(1-7). Thus, there is reciprocal down-regulation of the opposing peptide by the enzymes responsible for producing each peptide. Each peptide acts on its own distinct receptors (see white text boxes), with the Ang II type 1 receptor (AT1R) mediating the majority of the pro-hypertensive actions of Ang II and the Ang-(1-7) receptor activating pathways that serve as physiologic antagonists to the actions of Ang II. The actions of the AT2R are not yet clear, although several effects of PD123319 mimic the actions of Ang-(1-7). ACE, angiotensin converting enzyme; ACE2, angiotensin converting enzyme 2; NEP, neprilysin; AT1R, AT1 receptor; AT2R, AT2 receptor; AT(1-7) R, Ang-(1-7) receptor, also known as Mas; Sartans, AT1R antagonists, losartan class; PD123319, AT2R antagonist; D-Ala, D-Ala7-Ang-(1-7), the Ang-(1-7) receptor antagonist; PI3K, phosphoinositide 3-kinase; MAPK, mitogen-activated protein kinase.
- © American Society for Pharmacology and Experimental Theraputics 2008
References

Debra I. Diz, PhD, is Director of Basic Sciences Research, Hypertension & Vascular Research Center and a Professor in the Departments of General Surgery and of Physiology & Pharmacology, at Wake Forest University School of Medicine. Her education and training includes a BA from Randolph-Macon Woman’s College and a PhD in Pharmacology from the University of Tennessee Center for Health Sciences in Memphis. After a NIGMS Pharmacology Research Associate Fellowship at NIMH for post-doctoral training, she joined colleagues at the Cleveland Clinic Foundation. She is currently a Professor at Wake Forest University School of Medicine and is Chair of the Division of Pharmacology of the American Society of Pharmacology & Therapeutics. Dr. Diz’s research has identified the neuroanatomical and transmitter pathways subserving angiotensin II actions in brain areas involved in baroreceptor reflex control of the circulation. Long-standing studies investigate neural and hormonal interactions in the kidney and alterations in responsiveness to angiotensin peptides in renal ischemia, hypertension and most recently during aging. E-mail ddiz{at}wfubmc.edu; fax 336-716-2456.

