Visions of drug discovery
Cimetidine (Tagamet®)
One of the first drugs to be developed through rational drug design, cimetidine remains a stellar example of the power of pharmacology, not only to provide efficacious drugs, but also to reveal the basic underpinnings of human physiology in health and disease (1).

Discovery

By 1964, histamine was known to stimulate the secretion of stomach acid by a mechanism that was not inhibited by “antihistamines.” In order to target this novel mode of histamine agonism, a team of scientists at Smith Kline and French pioneered a project of rational drug design (see James Black’s reminiscence). Starting from the structure of histamine the only design lead they synthesized hundreds of compounds. The team’s implicit goal, the medicinal control of acid secretion, was framed within a grander pharmacological quest, largely unexplored at that time: proof of multiple receptor subtypes. The SK&F scientists would, over the course of time, prevail in this quest by discovering the basis for antihistamine-resistant gastric acid secretion: the H2 histamine receptor. The team’s first triumph lay in recognizing the histamine analog Nα-guanylhistamine as a partial, albeit nonspecific, agonist. From this lead, burimamide was developed, which was 100-fold more potent than Nα-guanylhistamine in inhibiting histamine activity, specifically at the novel (H2 subtype) histamine receptor. Burimamide was not satisfactory for oral administration, but another analog, metiamide, reached clinical assessment. Metiamide was toxic but instructive: replacement of its thiourea moiety with a cyanoguanidine group resulted in cimetidine (2).
James Black Reminisces

In 1948, Raymond Ahlquist published his landmark paper that hypothesized two kinds of receptors for adrenalin:αandβ. His hypothesis, essentially, was based on the invention, in 1940, of isoproterenol, whereby the terminal N-methyl group of adrenalin was replaced by an N-isopropyl group. Isoproterenol was a potent bronchodilator and cardiac stimulant, neither of which actions could be blocked by known “anti-adrenalin” drugs. Ahlquist posited isoproterenol as a selective stimulant of adrenalin β -receptors, whereas typical “anti-adrenalin” drugs defined the α receptors.
In 1958, I hoped to develop selectiveβ-receptor antagonists to control heart rate and rhythm. By analogy to isoproterenol, I assumed that selective antagonists might be found by modifying the terminal N-methyl group of adrenalin. But in that same year, Powell and Slater, hoping to make a long-acting bronchodilator, published the pharmacology of dichloroisoproterenol (DCI), in which the ring hydroxyl groups were replaced by chlorine atoms. Their hopes were not fulfilled; DCI was inactive as a bronchodilator but they noted that DCI exposure precluded the bronchodilator activity of isoproterenol. We showed that DCI was a very potent partial agonist on the cardiac pacemaker, my target tissue. Stephenson, my chemical colleague, immediately recognized that naphthalene had the same steric characteristics as DCI but quite different electronic features; in 1960, his synthesis of the β-naphthyl analog of isoproterenol resulted in the first selective β blocker.’
Now, turning to another of my target tissues at that time, I had published, in the 1950s, the effects of 5-hydroxytryptamine on histamine-stimulated gastric acid secretion. In those studies, I learned that antihistamines did not block this clinically important effect of histamine. The question in this context, raised by our invention ofβ-adrenergic blockers, was, Might histamine, like adrenalin, have its own “βreceptors”? At the time we asked that question, there were no known selective antagonists of acid secretion. Graham Durant, in a heroic bout of medicinal chemistry, replaced every available H atom of histamine with a methyl group; the relative agonist activity of these compounds was measured by Michael Parsons. He utilized in vitro assays of guinea pig ileum contraction (antihistamine sensitive) and guinea pig gastric acid secretion (antihistamine insensitive). The exciting result was that 4-methylhistamine proved to be fortyfold more selective for acid secretion than for ileal contractions, providing strong evidence for a second subtype of histamine receptors! We planned a program to make selective antagonists of this newly indicated second subtype (i.e., conceived as aβtype’) of histamine receptor. In this endeavor, we began to pursue a second analogy: having found that side-chain substitution controlled the agonist selectivity of adrenalinβ- blockers, whereas ring substitution produced antagonist selectivity, we hoped to determine whether selective histamine antagonism could be similarly achieved by modifying the side-chain moiety. Initial compounds produced by alkyl variations of the terminal amino group of 4-methylhistamine and replacing the amino group with a guanidine function produced potent selective agonists. We then put much effort into synthesizing new imidazoline analogs, but without success. Our synthetic work, however, did not prove to be completely unproductive, a happy circumstance that became apparent as certain assays were adjusted. In particular, we replaced the guineapig isolated stomach preparation with the guineapig right atrial preparation, and improved our quantitative design. To calibrate the new assay, Parsons turned back to our previously synthesized and assayed compounds. To our surprise, one of our earliest compounds, the guanidino analog of histamine, produced some inhibition of maximal histamine responses, the hallmark of a partial agonist! From there, Robin Ganellin systemically explored chain length (butyl was found optimal) and electronic features of side-chain and terminal groups, and he ended up with cimetidine! With H.O. Schild having already introduced the term H1for the antihistamine sensitive responses, we published the data of our ‘in-houseβreceptors’ in 1972, referring to specific antagonism of histamine H2receptors.’”
Indicated uses
-
acute duodenal ulcer
-
maintenance therapy after duodenal ulcer
-
acute benign gastric ulcer
-
erosive gastroesophageal reflux disease (GERD)
-
prevention of upper gastrointestinal bleeding
-
pathological hypersecretory conditions (i.e., Zollinger-Ellison Syndrome, systemic mastocytosis, and multiple endocrine adenomas)
Action and adversity
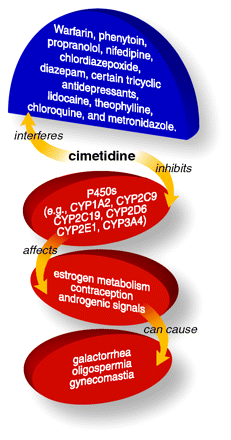
Cimetidine competitively inhibits histamine at H2 receptors of parietal cells and thus reduces gastric acid secretion. Cimetidine inhibits many cytochrome P450 isozymes, thereby delaying the elimination of certain drugs and ingested xenobiotics. Through its effect on stomach acid, cimetidine administration can interfere with drugs that are absorbed in a pH-dependent manner (e.g., ketoconazole).
Ins and outs
-
rapidly absorbed (oral); peaks at 45–90 min; t½ ~ 2 h
-
dose of 300 mg provides 80% inhibition of basal acid secretion
-
urine is principal route for excretion
-
24 h after oral dose, 48% of parent compound occurs in urine
-
24 h after parenteral dose, 75% of parent compound occurs in urine
-
major metabolite is sulfoxide derivative

On the market
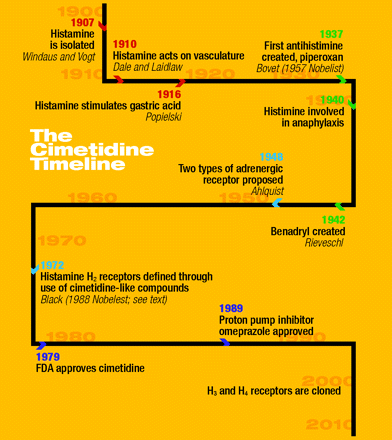
Cimetidine was the original blockbuster, reaching more than $1 billion sales in one year. It was first marketed in the UK in 1976, taking twelve years from the initiation of the H2-receptor antagonist program to commercialization. Development of longer-acting H2-receptor antagonists with reduced side effects, such as ranitidine, marked the downfall of cimetidine (3). (See the timeline.)
Footnotes
- © American Society for Pharmacology and Experimental Theraputics 2009

