Repression of Aryl Hydrocarbon Receptor Transcriptional Activity by Epidermal Growth Factor
The Aryl Hydrocarbon Receptor (AHR) mediates most if not all the many toxicological effects of the environmental pollutant 2,3,7,8-tetrachlorodibenzo-p-dioxin [(TCDD) or dioxin]. Dioxin elicits a wide variety of toxiocological effects in experimental animals, including immunotoxicity, teratogenesis, endocrine disruption, and multi-organ carcinogenesis (1). Dioxin is the most potent of a large number of industrial-era halogenated polyaromatic hydrocarbon pollutants, including other dibenzo-p-dioxins, dibenzo-p-furans and certain polychlorinated biphenyls. The AHR binds dioxin with high affinity, and experiments with AHR knockout mice indicate that all the toxicological effects of the compound are mediated by AHR (2, 3).
In the absence of ligand, AHR is located in the cytoplasm in a complex with two molecules of the 90-kDa heat shock protein (hsp90), the co-chaperone p23, and the immunophilin-like XAP2 protein [also known as AHR interacting protein (AIP)]. In the “classical” pathway of AHR action, after binding ligand, AHR translocates into the nucleus, discards its chaperone proteins, and dimerizes with the Aryl Hydrocarbon Receptor Nuclear Translocator (ARNT) protein, which, like AHR, is a member of the basic helix-loop-helix-Period-Arnt-Single-minded (bHLH-PAS) family of transcription factors. The AHR-ARNT dimer then binds to specific regulatory sequences, termed Xenobiotic Responsive Elements (XREs), in the upstream “enhancer” regions of responsive genes, the most thoroughly studied of which is the cytochrome P4501A1 (CYP1A1) gene. The AHR/ARNT dimer then recruits a number of coactivator proteins to the enhancer and promoter regions of the genes, which catalyze covalent modifications of histone proteins and alterations in nucleosomal structure in the immediate vicinity of the dimer, leading to the recruitment of RNA polymerase II and other general transcription factors to the promoter, resulting in the initiation of transcription of the gene (4, 5) (Figure 1⇓). The various toxic effects of dioxin were generally envisaged to result from the action of the products of genes, not yet necessarily identified, whose transcription was modulated by dioxin in this way.
More recently, however, other “non-classical” mechanisms whereby AHR mediates effects of dioxin have been described. For example, AHR cross-talks with Estrogen Receptor α (ERα). The negative effects of AHR on ER signaling appears to occur by several mechanisms, including: 1) inhibition of estradiol-induced expression of responsive genes by direct interaction of the AHR-ARNT complex with inhibitory dioxin responsive elements, resulting in disruption of interactions between proteins that bind DNA elements required for ER action and the basal transcription machinery; 2) synthesis of an unknown inhibitory protein; 3) competition for shared transcription factors, including ARNT; and 4) increased proteasomal degradation of ER (6, 7). Intriguingly, AHR ligands also reportedly induce the recruitment of the AHR-ARNT dimer to estrogen response elements (EREs) in ER-responsive genes. In this case, the AHR-ARNT complex does not directly contact DNA, but relies on the ER to do so. The AHR-ARNT complex appears to either activate or attenuate transcription of the ER-responsive genes, depending on the absence or presence of estradiol, respectively (8). It should be noted, however, that this model has been challenged (9). Recently, liganded AHR has been shown, in conjunction with ARNT, to direct the degradation of ERs and androgen receptors through its activity as a component of a novel ubiquitin-ligase complex (10). Liganded AHR also binds the retinoblastoma protein (Rb), thereby inhibiting cell-cycle progression independently of its binding to DNA (11). Liganded AHR can also bind the p65 subunit of Nuclear Factor Kappa Light Chain Enhancer of Activated β Cells (NF-κB) and thereby either suppresses or activates (depending on cellular context) the expression of NF-κB-dependent genes (12). NF-κB activation can reciprocally repress the transcriptional activity of AHR-ARNT, and a number of other examples are known in which regulatory pathways impact AHR activity. Sutter and coworkers recently described a novel such pathway (13). They showed that Epidermal Growth Factor (EGF) represses (89–99%) the dioxin-mediated induction of CYP1A1 in normal human keratinocytes. EGF-mediated repression arises from inhibiting the recruitment of the p300 coactivator [which is known to be required for dioxin-induction of CYP1A1 (14)] to the enhancer region of the CYP1A1 gene. The amount of intracellular p300 protein was not affected. Sutter and coworkers proposed two possible explanations for the effect of EGF. First, EGF treatment is known to increase the transcription of a large number of genes. Activation of some or all of these genes may involve recruitment of p300, thereby reducing the availability of the coactivator for CYP1A1 induction (i.e., “squelching”). Second, EGF receptor (EGFR) signaling may cause a posttranslational modification of p300, preventing it from associating with AHR-ARNT. Clearly, other explanations are also possible. Both models proposed by Sutter and coworkers would be expected to be relatively non-specific, because expression of many genes requires p300. It would therefore be interesting to investigate whether EGF inhibits expression of other genes in human keratinocytes. For example, does EGF impact the induction of glucocorticoid-responsive genes in these cells? Another question is whether EGF affects the induction of CYP1A1 (and other dioxin-responsive genes) in other cell types that express EGFR.
The affinity of dioxin for human AHR is about tenfold less than that for mouse AHR, and as a consequence, humans may be relatively resistant to the toxic effects of dioxin (15). Although dioxin has been classified as a group 1 carcinogen (i.e., carcinogenic to humans) by the International Agency for Research on Cancer, the only unequivocal toxic effect of the compound in humans is chloracne. Dioxin accelerates the differentiation of cultured normal human keratinocytes, and elicits some other features reminiscent of chloracne in these cells, including the development of cornified envelopes. Sutter and coworkers showed that EGF inhibits these processes in keratinocytes, and obtained evidence that these effects of EGF were mediated by EGFR. They also found that EGF administration decreased the expression of several dioxin-inducible genes involved in the later stages of keratinocytes differentiation. The implications of these studies is that EGF may inhibit the development of chloracne by reducing the recruitment of p300 to dioxin-inducible genes involved in the differentiation of keratinocytes and the development of chloracne.
A single dose of dioxin can cause very-long-term chloracne, probably reflecting the long half-life of dioxin (i.e., several years) in the human (16). Although some improvements in chloracne can occur within a few years (Figure 2⇓), the time to full recovery can extend to decades (17). The studies of Sutter and coworkers suggest that EGF could represent an effective treatment for chloracne, although it may be prudent to test the compound in an animal model of chloracne, such as the homozygous hairless (hr/hr) mouse (18), before applying the treatment in humans.
In conclusion, the work of Sutter and coworkers point to a potential therapeutic strategy for chloracne and provides a clear example of a mechanism whereby an agent negatively affects AHR-dependent induction of gene transcription by inhibiting the recruitment of a transcriptional coactivator.
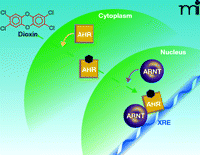
“Classical” pathway of AHR action. Dioxin (also denoted by the black hexagon) passes into the cytoplasm passively and binds to AHR. AHR translocates into the nucleus where it discards its associated chaperone proteins (not illustrated), dimerizes with ARNT, and binds to XRE sequences in responsive genes.

The effects of dioxin poisoning on the skin. Photographs of Victor Yuschenko before (left), immediately after he was poisoned by dioxin in 2004 (center), and in 2007 (right), showing the severity of his chloracne in 2004, and his partial recovery three years later. The photographs were obtained from a Ukrainian news blog. http://blog.kievukraine.info/2007/12/viktor-yushchenko-shows-arresting.html
Acknowledgments
I thank Kelly Joiner and Parrisa Solaimani for their help with this manuscript.
- © American Society for Pharmacology and Experimental Theraputics 2009
References

Oliver Hankinson, PhD, is a Professor in the Department of Pathology and Laboratory Medicine, and Director of the Molecular Toxicology Interdepartmental Doctoral Program at the University of California, Los Angeles (UCLA). He obtained his doctoral degree in Genetics from the University of Cambridge, England, and did postdoctoral research at Harvard; the University of Colorado, Denver; and the University of California, Berkeley. He joined the UCLA faculty in 1979. His research focuses on the mechanism of transcriptional activation by AHR and Hypoxia Inducible Factor (HIF), the mechanism(s) of carcinogenesis by polycyclic aromatic hydrocarbons, and the role of cytochrome P4502S1 (CYP2S1) in disease. For example, his group cloned the ARNT gene and delineated the role of ARNT in the AHR and HIF pathways. E-mail: ohank{at}mednet.ucla.edu; fax 310-794-9272.

