|
 
 |
|
CASE REPORT |
|
|
|
| Year : 2013 | Volume
: 19
| Issue : 4 | Page : 475-478 |
| |
Molecular characterization in a case of isolated growth hormone deficiency and further prenatal diagnosis of an unborn sibling
Ruchi Nadar1, Kavita Khatod2, Nikhil Phadke2, Chaitanya Datar3, Sujata Vaidya3, Anuradha Khadilkar4, Vaman Khadilkar1
1 Hirabai Cowasji Jehangir Medical Research Institute, Jehangir Hospital, Camp, Pune, Maharashtra, India
2 GenePath Dx, Causeway Healthcare, Phadke Hospital, Shivaji Nagar, Pune, Maharashtra, India
3 Sahyadri Medical Genetics and Tissue Engineering Facility, Shivajinagar, Pune, Maharashtra, India
4 Hirabai Cowasji Jehangir Medical Research Institute, Jehangir Hospital, Camp; GenePath Dx, Causeway Healthcare, Phadke Hospital, Shivaji Nagar, Pune, Maharashtra, India
| Date of Web Publication | 4-Jan-2014 |
Correspondence Address:
Anuradha Khadilkar
Cowasji Jehangir Medical Research Institute, Lower ground floor, Block V, Jehangir Hospital, 32, Sassoon Road, Pune-411 001, Maharashtra
India
 Source of Support: None, Conflict of Interest: None
DOI: 10.4103/0971-6866.124380

 Abstract Abstract | | |
Familial isolated growth hormone deficiency (GHD) type 1 is characterized by an autosomal recessive pattern of inheritance with varying degrees of phenotypic severity. We report a proband, with isolated GHD (IGHD) with very early growth arrest and undetectable levels of GH. Homozygous complete deletion of the GH1 gene was identified by real-time/quantitative polymerase chain reaction (RT/q-PCR) and confirmed by an independent molecular genetic method; the multiplex ligation-dependent probe amplification (MLPA) technique. Prenatal diagnosis was offered for the subsequent pregnancy in the mother of our proband. Identical heterozygous deletion of the GH1 gene was detected in both parents. The fetus had a similar homozygous deletion of the GH1 gene. We thus report a unique case with a confirmed mutation in GH1 gene in the proband followed by prenatal detection of the same mutation in the amniotic fluid which to our knowledge hitherto has not been documented from India.
Keywords: Antenatal diagnosis, GH1 deletion, isolated growth hormone deficiency
How to cite this article:
Nadar R, Khatod K, Phadke N, Datar C, Vaidya S, Khadilkar A, Khadilkar V. Molecular characterization in a case of isolated growth hormone deficiency and further prenatal diagnosis of an unborn sibling. Indian J Hum Genet 2013;19:475-8 |
How to cite this URL:
Nadar R, Khatod K, Phadke N, Datar C, Vaidya S, Khadilkar A, Khadilkar V. Molecular characterization in a case of isolated growth hormone deficiency and further prenatal diagnosis of an unborn sibling. Indian J Hum Genet [serial online] 2013 [cited 2016 May 24];19:475-8. Available from: http://www.ijhg.com/text.asp?2013/19/4/475/124380 |
| Introduction | |  |
Isolated growth hormone deficiency (IGHD) has an incidence of 1:4,000 to 1:10,000 live births. Only 11-18% of these cases have an identifiable mutation and familial occurrence has been reported in 3-30% patients. [1],[2] Having an autosomal recessive inheritance, IGHD type 1 is more common in consanguineous marriages. Some genetic variants of IGHD are associated with a phenotype of evolving multiple pituitary hormone deficiency. [3] It is useful to have a genetic diagnosis to get an insight into the expected response to GH therapy and to determine if any other endocrine abnormalities are likely to occur as a part of the spectrum of the disorder. It is also necessary to know the mutation status in the proband and parents to determine the exact mode of inheritance. Phenotypic severity and poor response to therapy associated with some genetic variants of IGHD thus justifies prenatal diagnostic workup of the fetus in a high-risk pregnancy.
Thus, objective of our study was to detect if there was a prenatally diagnosable genetic cause of IGHD in the unborn sibling of a child who was clinically GHD. Specific aims were to: (i) Identify genetic cause of GHD in the (proband). (ii) If detected in the proband, determine if it was inherited from one/both parents. (iii) To check for presence of same in amniotic fluid.
| Case Report | | |
The proband had presented at 1 year with severe short stature. He was born of a consanguineous marriage and belonged to an inbred Muslim family. Frontal bossing, dysmorphic face with hypoplastic features, micropenis, and severe but proportionate short stature (height for age z score - 9.6) [4] were suggestive of GHD. Clonidine stimulated GH concentrations were undetectable (<0.1 ng/mL). Response to GH therapy was excellent and at present he continues to take GH.
Parents of the proband presented for prenatal diagnosis for their next child at 13-14 weeks of gestation. After a proper pretest counseling and patient consent, amniocentesis procedure was performed at 16 weeks to obtain the amniotic fluid sample for molecular genetic testing.
Molecular genetic methods
Since a clinical diagnosis of IGHD had been made, known genes for IGHD for which testing was available were assessed, that is, a total of 17 amplicons of the GH releasing hormone-receptor (GHRHR) and GH1 genes were tested. Vitamin D receptor (VDR) gene was used as a control. Real-time/quantitative polymerase chain reaction (RT/q-PCR) analysis which looks for changes in copy numbers in the patient compared to a control sample was carried out on genomic DNA of proband extracted from 200 μL of peripheral blood (Bioneer kit, Korea).
DNA was further tested using an independent molecular method, multiplex ligation-dependent probe amplification (MLPA). MLPA analysis was performed using the SALSA MLPA P216-A2 GHD Probemix (MRC-Holland, Netherlands) as per the manufacturer's instructions. The mix included MLPA probes spanning the GH1, PROP1, POU1F1, GHRHR, HESX1, LHX3, LHX4 genes. Similar analysis was also carried out on the parents' samples.
Ten milliliter of the amniotic fluid sample was resuspended in 250 μl of phosphate buffered saline. Genomic DNA was isolated and used for analysis of the GH1 gene by RT/q-PCR and MLPA (as described above).
| Results | | |
RT/q-PCR followed by gel electrophoretic analysis in the proband showed absence of amplification of all five GH1 exons; however, expected amplification was seen in healthy control [Figure 1]a and b, suggesting a homozygous deletion of all exons of the gene.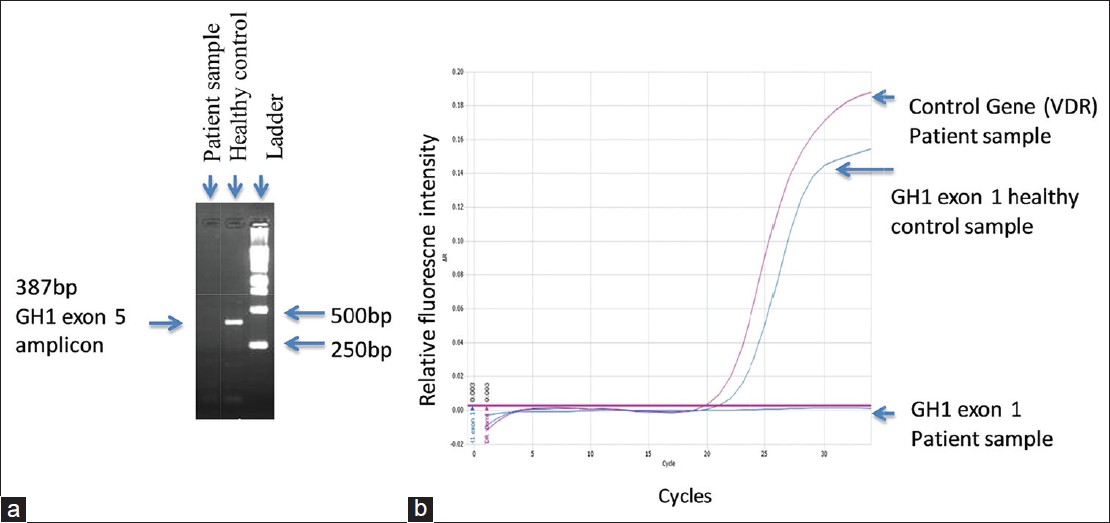 | Figure 1: (a) Gel electrophoretic analysis of polymerase chain reaction (PCR) products of growth hormone (GH) 1 exon 5: Expected amplification is seen in healthy control. There was no amplification in the proband. (b) Real-time SYBR Green PCR of GH1 exon 1. Healthy control showed expected amplification. No amplification was seen in the proband's sample. Amplification of the VDR gene used as the control gene in the patient's sample showed normal amplification
Click here to view |
RT/q-PCR specificity was confirmed by melt curve analysis (data not shown).
MLPA analysis [Figure 2] showed complete lack of GH1 amplification in the proband; whereas, it was in reduced intensity in both parents.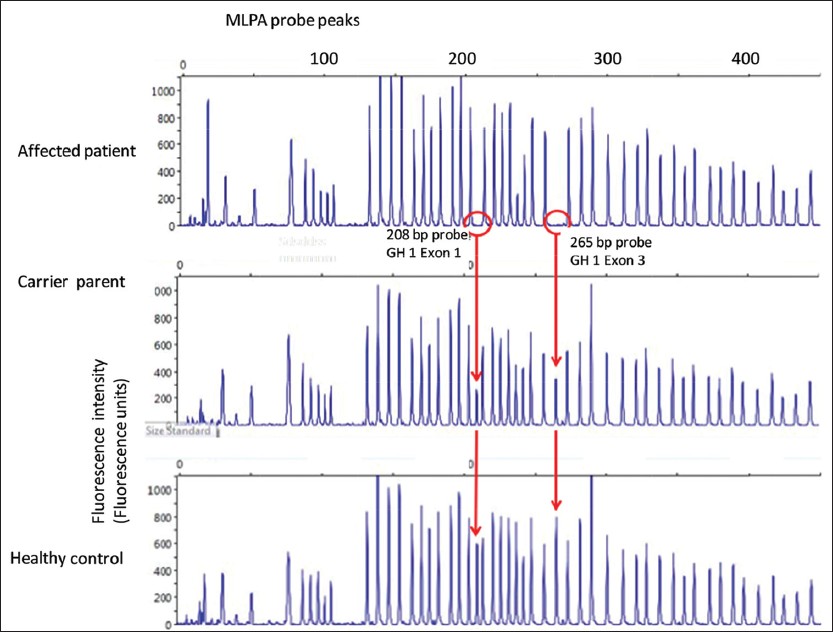 | Figure 2: Multiplex ligation-dependent probe amplification (MLPA) analysis of proband sample along with father's sample (maternal data not shown). The GH1 specific amplicons (208 base pairs (bp) and 265 bp) are absent in the proband samples, at reduced intensity (nearly half normal indicating a heterozygous state) in the parental sample, and at full intensity in the control sample from a healthy male. (MLPA analysis of fetus not demonstrated graphically)
Click here to view |
ΔCt values obtained on PCR analysis showed very low amplification in the proband, suggesting a complete deletion of GH1 gene. Whereas, data of parents suggested a heterozygous deletion [Table 1].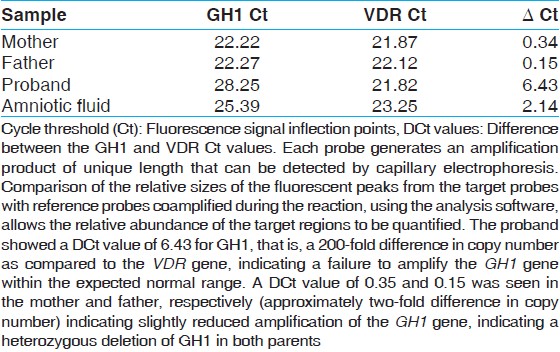 | Table 1: Quantitative (real-time) SYBR green polymerase PCR on growth hormone 1 gene and the human vitamin D receptor gene
Click here to view |
Amniotic fluid sample also showed markedly reduced copy numbers of the GH1 gene on RT/q-PCR; suggesting complete deletion of gene in the fetus. ΔCt values were similar to that of the proband (Figure not shown, data in [Table 1]. Findings were independently confirmed by MLPA. All test controls (positive and negative) performed within expected range.
| Discussion | | |
We report here genetic analysis of a family in which proband had a homologous complete deletion of GH1 gene (demonstrated by RT/q-PCR, confirmed by gel electrophoresis and MLPA), manifested at an early age, had severe phenotype of GHD, and showed good response to GH therapy. Parents were heterozygous carriers of the same deletion suggesting autosomal recessive inheritance [Figure 3].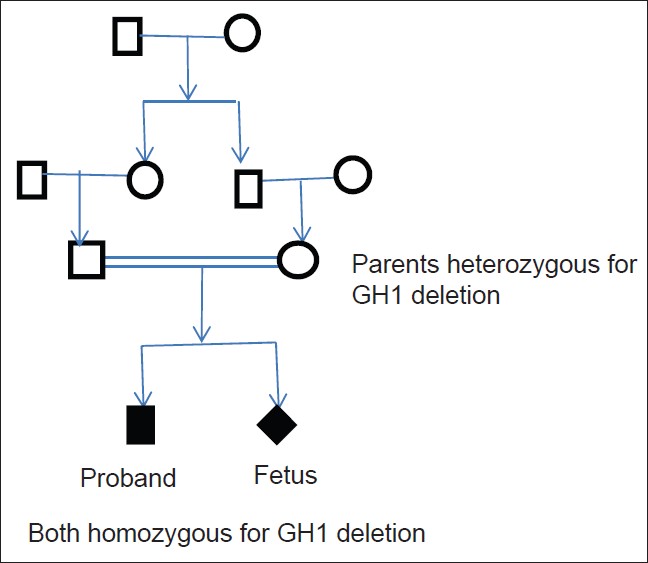 | Figure 3: Family tree both parents are heterozyogus carriers of the GH 1 deletion. The proband and unborn fetus are homozygous for the same deletion
Click here to view |
Both parents are heterozygous carriers of the GH1 deletion. The proband and unborn fetus is homozygous for the same deletion.
Our data suggest that the fetus also had homozygous deletion of the GH1 gene. All PCR and MLPA data were in concordance. MLPA is an extension of PCR that allows highly multiplexed detection of relative copy number variations of target sequences in a fast, reliable, and cost-effective manner. [5]
Mutations in patients with GHD are more likely to be identified in familial cases (34%) and in patients with a height Z score ≤ -4.5 (20%). [1],[6],[7] Genetic testing is thus strongly indicated in consanguineous marriages, those with positive family history and severe phenotype. GH1 deletions result in complete block in GH synthesis, hence the phenotype is severe and anti-GH antibodies develop during recombinant GH therapy. [8] Mullis and Brickell have reported antenatal diagnosis of two cases of GH1 deletions in two at-risk pregnancies, using the PCR. [9] To the best of our knowledge, this is the first Indian report of antenatal diagnosis of GH1 deletion.
To conclude, we report a family where homozygous GH1 deletion was confirmed in a suspected case of IGHD followed by successful prenatal diagnosis for the subsequent unborn sibling of the proband. IGHD type 1 was confirmed by heterozygous status of both parents.
Treatment with GH is available, though still quite expensive in India. Therefore in these circumstances, antenatal diagnosis provides an opportunity for appropriate genetic counseling and early detection.
| References | | |
| 1. | Alatzoglou KS, Turton JP, Kelberman D, Clayton PE, Mehta A, Buchanan C, et al. Expanding the spectrum of mutations in GH 1 and GHRHR: Genetic screening in a large cohort of patients with congenital isolated growth hormone deficiency. J Clin Endocrinol Metab 2009;94:3191-9. 
|
| 2. | Vimpani GV, Vimpani AF, Lidgard GP, Cameron EH, Farquhar JW. Prevalence of severe growth hormone deficiency. Br Med J 1977;2:427-30.
|
| 3. | Alatzoglou KS, Dattani MT. Genetic causes and treatment of isolated growth hormone deficiency-an update. Nat Rev Endocrinol 2010;6:562-76.
|
| 4. | WHO Multicentre Growth Reference Study Group. WHO Child Growth Standards based on length/height, weight and age. Acta Paediatr Suppl 2006;450:76-85.
|
| 5. | Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res 2002;30:e57.
|
| 6. | Mullis PE, Akinci A, Kanaka C, Eblé A, Brook CG. Prevalence of human growth hormone-1 gene deletions among patients with isolated growth hormone deficiency from different populations. Pediatr Res 1992;31:532-4.
|
| 7. | Desai MP, Mithbawkar SM, Upadhye PS, Shalia KK. Growth hormone (GH-1) gene deletions in children with isolated growth hormone deficiency (IGHD). Indian J Pediatr 2012;79:875-83.
|
| 8. | Rosenfeld RG, Cohen P. Disorders of growth hormone/Insulin like growth factor secretion and action. In: Sperling MA, editor. Pediatric Endocrinology. 3 rd ed. Saunders: Elsevier; 2008. p. 254-334.
|
| 9. | Mullis PE, Brickell PM. The use of the polymerase chain reaction in prenatal diagnosis of growth hormone gene deletions. Clin Endocrinol 1992;37:89-95.
|
[Figure 1], [Figure 2], [Figure 3]
[Table 1]
|