|
|
An Uncommon Cause of Peripheral Neuropathy
Naushira Pandya, MD;
Mark Byler, MD;
Scott Armistead, MD
Arch Fam Med. 1998;7:85-87.
ABSTRACT
The following case report is of a middle aged man with a progressive neurologic disability who was brought to our emergency department. Caregivers could no longer manage him once he lost the ability to ambulate and became incontinent. At that time his neurologic and hematologic parameters were fairly typical of cobalamin deficiency. We are reporting this case to emphasize that cobalamin deficiency is not always due to pernicious anemia in the elderly or the malnourished alcoholic. Deficiency from uncommon causes can manifest at any age and result in severe morbidity.
INTRODUCTION
Cobalamin deficiency is usually seen in the elderly secondary to pernicious anemia. Its neurologic manifestations usually start as dorsal column degeneration with paresthesias, impaired vibrator sense, and loss of joint position sense. If the deficiency is not treated, progressive involvement of motor neurons leads to impaired mobility with increased disability and uncertain recovery.
In this case report, we are describing a man who presented with neurologic symptoms of cobalamin deficiency starting in his late thirties; however, his condition was not diagnosed until 6 years later. During this time his symptoms prgressed from paresthesias of his hands to loss of his ability to walk. Despite treatment, our patient experienced only a partial recovery.
PATIENT REPORT
A 45-year-old man was admitted from the emergency department in June 1992 complaining of inability to walk, incontinence of both bladder and bowel, and erectile dysfunction. Two weeks before admission he had been able to ambulate with a walker.
In 1986 he had begun to develop paresthesias of the hands, first noted while manipulating the controls of his camera. He waited 2 years before seeking medical attention and, in 1988, was referred to a neurologist by his family physician. By that time, he had also developed paresthesias and burning in both hands and feet, leg cramps, and stumbling. Neurologic examination at that time was significant for bilateral weakened hand grips, atrophy of the interossei, hyporeflexia, and reduced distal sensation to touch, pinprick, and vibration distal to the wrists and ankles.
Initial evaluation performed in 1988 included the following: (1) electromyography, which showed delayed conduction, mostly in the sensory fibers, with diffuse, distal denervation changes in all extremities; (2) magnetic resonance imaging of the head, the results of which were normal; (3) creatine kinase, normal; (4) antinuclear antibody, normal; (5) serum protein electrophoresis, normal; (6) urine screen for heavy metals, negative; (7) chest radiograph, normal; (8) erythrocyte sedimentation rate, 42 mm/h; and (9) cerebrospinal fluid 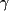 -globulins, increased at 15%, suggestive of an inflammatory central nervous system process. -globulins, increased at 15%, suggestive of an inflammatory central nervous system process.
Progressive polyradicular neuropathy was diagnosed, and initial treatment was undertaken with a trial of low-dose (50 mg) amitriptyline daily followed by high-dose corticosteroids (dose unknown), without improvement.
Four years later, in 1992, the patient came to our emergency department with progressive deterioration in all activities of daily living to the extent that his caretakers could no longer care for him. On physical examination, the patient was a well-developed and well-nourished man. Results of cardiovascular, pulmonary, abdominal, and testicular examinations were normal. The bladder was not palpable, nor was there any lymphadenopathy. Neurologic examination showed normal mental status, normal optic fundi, and absent fasciculations, tremor, or clonus. Interosseous atrophy was present and more prominent in the right hand. Upper-extremity motor strength and coordination were normal. Examination of thelower extremities was significant for diminished muscle tone, with 4/5 strength in the proximal musculature and 3/5 strength in the distal musculature. Coordination was poor in the lower extremities, as manifested by heel to shin testing. Joint position sense was absent in the lower extremities, as was vibratory sense up to the level of T-4. Sensation to light touch, pain, and temperature was also absent to T-4. Patellar and Achilles reflexes were absent, but positive crossed adductor reflexes and Babinski sign were present bilaterally. Rectal examination showed diminished tone.
Initial laboratory values are shown in Table 1. In view of the patient's marked peripheral neuropathy, sensory level, loss of dorsal column function, and macrocytosis, further investigations were ordered (Table 2).
|
|
|
|
Table 1. Initial Laboratory Values*
|
|
|
|
|
|
|
Table 2. Results of Additional Investigations*
|
|
|
The differential Schilling test was performed because of the marked macrocytosis and subnormal cobalamin levels. The patient did not absorb radioactively labeled cobalamin without intrinsic factor (stage I) or with intrinsic factor (stage II). After receiving a 10-day course of oral tetracycline, 250 mg 4 times a day, to treat possible small-bowel overgrowth, the patient absorbed and excreted cobalt 59– and 58–labeled cobalamin normally.
The patient was initially treated with intramuscular cobalamin and received a total of 4000 mg during a 2-week period. He was transferred to the rehabilitation unit, and within 3 months he had regained some sensation in the abdominal region. Truncal stability returned, and the patient was able to stand without assistance. Parts I and II of the Schilling test excluded pernicious anemia as a cause of the patient's condition. Because cobalamin absorption normalized after a 10-day course of antibiotic, bacterial overgrowth was presumptively diagnosed. Enteroclysis was performed to search for both blind loop syndrome and hypomotility, but results were normal. Malabsorption was considered, but the patient had no history of weight loss or steatorrhea. Absorption of orally administered D-xylose was normal.
On repeated examination 1 year later, in July 1993, vibratory, light touch, pain, and temperature sensation had improved, moving from the T-4 level to T-12–L-1 for vibratory, pain, and light touch sensation and L-5 for temperature. Deep tendon reflexes remained unchanged at 2/4 to upper extremities, 0/4 to Achilles tendon, and 0/4 to patella. The patient had regained truncal stability and was able to sit without assistance. He was able to stand and walk a few steps with a walker. He remained incontinent of bladder and bowel. During the next few years, the patient did not gain further clinical improvement.
COMMENT
The hematologic measures and neurologic manifestations of our patient's disease are fairly typical of cobalamin deficiency. His case is of particular interest because of his youth, the delay in diagnosis, and the uncommon cause of his cobalamin deficiency.
In early cobalamin deficiency, the hematologic profile may be normal. Neurologic symptoms occur in the absence of hematologic abnormalities in 25% to 28% of cases.1 The hematologic findings of cobalamin deficiency may also be masked in patients who are taking supplementary folate. Our patient's initial evaluation in 1988 apparently did not include a complete blood cell count, so it is unclear whether hematologic manifestations were present at that time.
Our patient displayed a variety of neurologic manifestations characteristic of cobalamin deficiency. His paresthesias, impaired vibratory sense, and loss of joint position sense indicated dorsal column involvement. Lower-extremity weakness, crossed adductor reflexes, and Babinski sign suggest corticospinal tract involvement. The spinothalamic tract was also affected, as manifested by the sensory level on his trunk. Peripheral nerve involvement was suggested by distal symmetrical loss of superficial sensation and the absence of patellar and Achilles reflexes. All of these findings are fairly typical of subacute combined degeneration of the cord with upper and lower neuron involvement.2-4
The patient's first symptoms, notably paresthesias in his hands, began in 1986, when he was in his late 30s. Cobalamin deficiency is most commonly seen in elderly patients, a factor contributing, perhaps, to the delay in diagnosis in our patient. Cobalamin stores in a normal individual last for approximately 33 to 45 months,5 so his deficiency probably began in the early 1980s. The diagnosis was not made, however, until 1992, by which time he had lost continence of both bowel and bladder and the ability to walk.
Pernicious anemia accounts for 65% to 75% of cobalamin deficiency in the United States.4 Cobalamin deficiency unrelated to pernicious anemia is rare and may result from a number of causes.6 These include deficiency with normal absorption (inadequate dietary intake, bacterial overgrowth, Diphyllobothrium latum [fish tapeworm] infection, total gastrectomy, and nitrous oxide exposure [dental personnel]), deficiency with malabsorption (drugs that interfere with absorption [aminosalicylic acid, neomycin, ethanol, colchicine, and biguanides [metformin]), Crohn disease, tropical sprue, and ileal resection), and inherited congenital disorders (defective intrinsic factor and transcobalamin II deficiency).
In our patient, bacterial overgrowth in the absence of a blind loop was the apparent cause of cobalamin deficiency. Small-bowel bacterial overgrowth competes for available cobalamin.7-10 Causes of this condition include partial bowel obstruction, hypomotility disorders, diabetes mellitus, irradiation, mesenteric ischemia, scleroderma, and gastric hypochlorhydria.5, 11-13 Bacterial overgrowth can be encountered in conditions of altered immunity (humoral response), such as diabetes mellitus, human immunodeficiency virus infection, cirrhosis, malnutrition, and hypogammaglobulinemia. In our patient these were largely excluded by history, physical examination, and laboratory tests. The C14-xylose breath test, which is highly sensitive and specific, can be used to confirm small-intestine bacterial overgrowth. We elected not to do this because of lack of availability in our institution and our patient's prompt response to tetracycline as manifested by normalization of absorption of labeled cobalamin.
Repeated laboratory studies done in April 1993 showed the following normal values: cobalamin, 419 pg/mL; hemoglobin, 160 g/L; hematocrit, 0.47; and mean corpuscular volume, 83.0 fL. Despite this prompt response, we elected to keep treating our patient with monthly cobalamin injections rather than just oral replacement. Our choice for injectable cobalamin treatment was 2-fold: it is inexpensive, easy to administer, and had proven efficacy; and although it would take years to deplete cobalamin stores once replaced, it seemed inappropriate to risk even marginal deficiency in the individual to prove our diagnosis of bacterial overgrowth. Gastric aspirate pH analysis can exclude gastric hypochlorhydria.
Primary care physicians are in a prime position to conduct systematic studies of patients with neurologic symptoms. Early diagnosis of cobalamin deficiency not only can be cost-effective but can prevent considerable morbidity. Checking a cobalamin serum level or a urine test for homocysteine or methylmalonic acid is an important part of the evaluation of any patient with peripheral neuropathy or nonspecific neuropsychiatric symptoms.
AUTHOR INFORMATION
Accepted for publication July 3, 1997.
We are grateful to Diane Harper, MD, MPH, Dartmouth-Hitchcock Medical Center, Lebanon, NH, and Dave Rush, PharmD,Truman Medical Center-East, for their guidance, and to Gayle Kelley for manuscript assistance.
Reprints: Mark Byler, MD, Department of Community and Family Medicine, University of Missouri–Kansas City School of Medicine, 7900 Lee's Summit Rd, Kansas City, MO 64139.
From the Departments of Internal Medicine and Geriatrics, University of Missouri–Kansas City School of Medicine, and Department of Community and Family Medicine, Truman Medical Center-East, Kansas City, Mo (Drs Pandya and Byler); and Tri-County Medical Corp, Richmond, Va (Dr Armistead).
REFERENCES
1. Lindenbaum J, Healton EB, Savage DG, et al. Neuropsychiatric disorders caused by cobalamin deficiency in the absence of anemia or macrocytosis. N Engl J Med. 1988;318:720-728.
2. Diseases of the nervous system due to nutritional deficiency. In: Adams RD, Victor M, eds. Principles of Neurology. 4th ed. New York, NY: McGraw-Hill Book Co; 1989:820-845.
3. Shevell MI, Rosenblatt DS. The neurology of cobalamin. Can J Neurol Sci. 1992;19:472-486.
WEB OF SCIENCE
| PUBMED
4. Healton EB, Savage DG, Brust JC, Garrett TJ, Lindenbaum J. Neurologic aspects of cobalamin deficiency. Medicine. 1991;70:229-245.
PUBMED
5. Chesner IM, Montgomery RD. Small bowel contamination and vitamin B12 deficiency in the elderly. J Clin Gastroenterol. 1986;8:447-450.
PUBMED
6. Toskes PP, Deren JJ. Progress in gastroenterology. Gastroenterology. 1973;65:662-683.
PUBMED
7. Goldstein F. Mechanisms of malabsorption and malnutrition in the blind loop syndrome. Gastroenterology. 1971;61:780-784.
PUBMED
8. Schjonsby H. The mechanism of vitamin B12 malabsorption in blind-loop syndrome. Scand J Gastroenterol. 1973;8:97-99.
PUBMED
9. The blind-loop syndrome. Lancet. 1967;1:85-86. Editorial.
PUBMED
10. Giannella RA, Broitman SA, Zamcheck N. Competition between bacteria and intrinsic factor for vitamin B12: implications for vitamin B12 malabsorption in intestinal bacterial overgrowth. Gastroenterology. 1972;62:255-260.
PUBMED
11. Gracey M. Intestinal absorption in the ‘contaminated small-bowel syndrome.'. Gut. 1971;12:403-410.
FREE FULL TEXT
12. Banwell JG, Kistler LA, Giannella RA, Weber FL Jr, Lieber A, Powell DE. Small intestinal bacterial overgrowth syndrome. Gastroenterology. 1981;80:834-845.
PUBMED
13. Neale G, Gompertz D, Schonsby H, Tabaqchali S, Booth CC. The metabolic and nutritional consequences of bacterial overgrowth in the small intestine. Am J Clin Nutr. 1972;25:409-1417.
|