Selective Inhibitors of Cyclooxygenase-2
A Growing Class of Anti-Inflammatory Drugs
- Center for Experimental Therapeutics and
- *Department of Anesthesia University of Pennsylvania Philadelphia, USA
- Address correspondence to GAF. E-mail garret{at}spirit.gcrc.upenn.edu; fax 215-573-9135.
Abstract
For about two years, selective inhibitors of cyclooxygenase-2 have been hailed as powerful additions to the armamentarium of nonsteroidal anti-inflammatory drugs (NSAIDs). Predicted by financial analysts to become among the pharmaceutical industry's greatest-ever blockbusters, two so-called coxibs are now widely utilized clinically: rofecoxib (Vioxx, Merck) and celecoxib (Celebrex, Monsanto). The clinical advantages of the coxibs over traditional NSAIDs are related to the distinct roles played by cyclooxygenase-2 in comparison to its earlier described isoform, cyclooxygenase-1. Ongoing research into the disparate roles of the two isoforms will have implications not only for the pharmaceutical use of coxibs, but also for our basic appreciation of inflammation, cardiovascular diseases, Alzheimer's disease, cancer, and more.

Introduction
Prostaglandin G/H synthase catalyzes the transformation of arachidonic acid (AA) into the pivotal endoperoxide intermediate, prostaglandin H2 (PGH2), that serves as the substrate for the production of a variety of prostanoids with diverse and potent biological actions (Figure 1⇓). Prostaglandin G/H synthase is a bifunctional enzyme, possessing both the cyclooxygenase activity (actually, a bis-oxygenase activity) that converts AA into PGG2, as well as the peroxidase activity that reduces PGG2 to produce PGH2. The enzyme is commonly known by its trivial name, cyclooxygenase (COX).
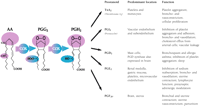
Prostanoid activities derived from COX activity. Prostaglandin G/H synthase (COX) is bifunctional: its bis-cyclooxygenase activity, directed both at C-11 and C-15 of arachidonic acid (AA), is indicated in purple; its hydroperoxidase activity, directed to the peroxide adduct at C-15 of prostaglandin G2 (PGG2), is indicated in blue.
COX inhibitors have been used medicinally for more than a century, although the biological basis for their efficacy has been appreciated only for a few decades. One of the earliest inhibitors to be used therapeutically was aspirin, which inhibits the enzyme's cyclooxygenase activity by irreversibly acetylating an active-site serine residue (1). Aspirin is thus distinctive among the structurally disparate members of the nonsteroidal anti-inflammatory drugs (NSAIDs) that otherwise function as competitive inhibitors of COX by reversibly occupying the binding site for AA.
Efforts to identify and develop novel NSAIDs were intensely invigorated about ten years ago with the discovery that COX exists as two distinct isoforms. Expression of the novel isoform, termed COX-2, proved to be upregulated by bacterial lipopolysaccharide (LPS), cytokines, growth factors, and tumor promoters, thereby distinguishing it from the primarily constitutively expressed COX-1 isoform (2-5).
Based on its distinctive expression profile, COX-2 was speculated to account for prostanoid formation in disease states such as inflammation and cancer. Thus, it was reasoned, the anti-inflammatory and analgesic properties of aspirin and other NSAIDs might be due specifically to the inhibition of the COX-2 isoform. Indeed, several researchers were able to show that the glucocorticoids—also a class of anti-inflammatory agents—could downregulate the induction of COX-2, amongst many other actions (3). In addition to its regulation at the level of transcription, COX-2 activity may be affected by cytokines, growth factors, and oncogenes that function to prolong COX-2 transcript half-life (6). COX-1 has the structural features of a constitutive gene, although its expression has recently also been found to be regulated in some settings. Indeed, the expression of both COX-1 and COX-2 is increased in inflamed joints and in atherosclerotic plaque (7, 8). The dichotomy of a “constitutive” COX-1, with solely “housekeeping” functions under physiological conditions, and an “inducible” COX-2, accounting for prostanoid production in disease states, is thus an oversimplification of biological reality.
The catalytic activities of COX-1 and COX-2, and their tertiary structures as delineated from crystallographic data, are remarkably similar (9). There is an important catalytic distinction, however, in that COX-2 manifests a broader substrate specificity that arises from a more accommodating hydrophobic tunnel leading to the active site. This tunnel may thus influence the spectrum, quantity, or duration of prostanoid production by COX-2 (10). The recognition of multiple COX isoforms has had one of the greatest impacts on the development of NSAIDs since the original synthesis of aspirin from salicylic acid more than a century ago.
Experimental Verification of COX Inhibitor Selectivity
The chemical synthesis of COX-2 inhibitors, or coxibs, necessitated specific assays of COX-1 and COX-2 inhibition in vitro. Screening for selective COX inhibitors has been facilitated by the use of purified recombinant human COX-1 and COX-2 isoforms in rapid and simple assays such as those that measure oxygen consumption. Such assays, however, do not recreate physiological conditions and thus cannot assess drug–enzyme interactions as they occur within the more complex biological milieu. For this reason, the differential inhibition of COX-1 and COX-2 activities in human blood has been increasingly relied upon during the past few years (11, 12). Platelet COX-1 activity may be assayed by measuring the production of thromboxane B2, a stable hydrolysis product of thromboxane A2 (Figure 1⇑) during whole blood clotting; LPS-induced COX-2 activity is determined as a function of PGE2 production by monocytes in heparinized plasma samples that have been pretreated with aspirin so as to obliterate platelet COX-1 activity. Through such assays, compounds can be evaluated for selective inhibitory activity either in vitro, by adding increasing concentrations of the inhibitor to whole blood, or ex vivo, by correlating variable circulating levels of the inhibitor (after oral dosing) with COX activity. Although the whole blood assays more closely mimic physiological conditions of COX activity, it should nevertheless be noted that even the most “clinically relevant” determination of inhibitor selectivity remains a function of the experimental concentrations of inhibitors and substrates. (For example, the selectivity of the currently available COX-2 inhibitors, as discussed below and elsewhere, is dose-dependent and may be lost at higher concentrations of inhibitor.)
Through the use of the in vitro whole blood assay, the concentration of celecoxib required to inhibit COX-1 or COX-2 by 50 percent (IC50) in human whole blood has been found to approximate that of two other NSAIDs (i.e., meloxicam and nimesulide) (13-16). Rofecoxib is the more biochemically selective COX-2 inhibitor of the two currently prescribed coxibs. Another coxib in development, valdecoxib, will approximate the selectivity of rofecoxib, and yet another, etoricoxib, appears to be approximately three times more selective than rofecoxib (Figure 2⇓) (17). It should be noted that although the traditional NSAIDs may show modest selectivity for a given COX isoform (IC50 ratios ranging between 0.5 and 20), in no such instance have comparative clinical trials (such as those performed for the coxibs, see below) or hospitalization analyses established correlations with drug efficacy and/or safety (18, 19). Thus, we feel that the description of traditional NSAIDs as “preferential” for COX-2 or COX-1 is potentially misleading.
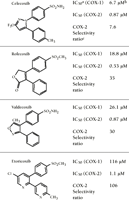
Structures and selectivity ratios of four coxibs as measured in human whole blood assays. In addition to the two COX-2 inhibitors that are currently on the market (i.e., celecoxib and rofecoxib), data are shown for two COX-2 inhibitors that are currently in clinical development (i.e., valdecoxib and etoricoxib).
a IC50 is the inhibitor concentration necessary to reduce COX rates by 50 percent.
b All data were obtained through the human whole blood assay (17).
c The selectivity of each compound for the COX-2 isoform is determined as the ratio IC50 (COX-1)/IC50 (COX-2).
The COX-2 Hypothesis
The “COX-2 hypothesis” proposes that at equally effective doses, selective COX-2 inhibitors will cause less serious gastrointestinal (GI) adverse effects than traditional nonselective NSAIDs. The effective clinical development of COX-2–selective inhibitors would thus rest on two assumptions: a) COX-2 inhibition is necessary and sufficient for analgesic/anti-inflammatory efficacy, and b) COX-1 inhibition is largely responsible for the serious GI toxicity of conventional NSAIDs. It must additionally be recognized that biochemical selectivity may represent only one of many factors that will influence the clinical outcome in any individual patient undergoing treatment with a COX-2 inhibitor.
The Role of COX-2 in Inflammation
NSAIDs are widely used for the treatment of arthritis and other conditions that call for general analgesia. Evidence derived from animal models of inflammation suggests that appropriate doses of rofecoxib and celecoxib achieve efficacy that is comparable to conventional NSAIDs (20, 21). The accurate assessment of the effectiveness of COX-2 inhibitors, however, may be complicated by the fact that pro- as well as anti-inflammatory prostanoids can be generated in some of the animal models and perhaps in humans (22). Although COX-2 appears to be the dominant source of prostaglandin formation in inflammation, there is some suggestion that both isoforms of the human enzyme may contribute to acute inflammatory responses (23).
The mouse models of COX gene deletion similarly exhibit some discrepancies with the COX-2 hypothesis. For example, both AA and the tumor promoter tetradecanoylphorbol acetate (TPA) induce COX-2 expression and cause skin inflammation in normal mice, but curiously, COX-2 knockout mice mount the same level of induced inflammation in response to AA and TPA (24). Moreover, the deletion of the COX-1 gene mitigates the inflammatory response to AA but not to TPA (25). These results do not necessarily impugn the COX-2 hypothesis, because the response of the COX-1 knockout mice to AA was scored early, possibly when neutrophilic COX-1 activity in wild-type animals would predominate over the COX-2 activity associated with monocytes. Similarly, because both COX isozymes may more generally influence the development of immunity (26, 27), the absence of either isozyme during development might influence adult mouse models of inflammation.
Gastrointestinal Toxicity and Inhibition of COX-2
The second assumption that is posited in the COX-2 hypothesis is that selective inhibitors of COX-2 will avoid the adverse GI effects related to COX-1 inhibition. Although 10 to 20 percent of patients taking NSAIDs experience dyspepsia (28, 29), serious problems with GI adverse effects are not frequent. Nevertheless, the large number of people taking NSAIDs makes the GI adverse effect profile a significant concern. About 5 to 15 percent of patients discontinue taking NSAIDs due to dyspepsia (30), and the annual mortality rate associated with GI effects of NSAIDs has been estimated at 0.22 percent (31).
Several studies have evaluated the efficacy and side effects of the new drugs, celecoxib and rofecoxib, in comparison to other NSAIDs. The Vioxx Gastrointestinal Outcomes Research (VIGOR) Trial is a prospective study of the incidence of GI perforation ulceration or bleeding (PUB) in rheumatoid arthritis (RA) patients that are administered rofecoxib versus naproxen (32). Significantly, the annual incidence of confirmed PUB is reduced by 54 percent in the rofecoxib group as compared to patients receiving naproxen (2.1 percent vs 4.5 percent). The Celecoxib Long-term Arthritis Safety Study (CLASS) combines two prospective trials that compare the adverse effects of celecoxib with those of reference NSAIDs (diclofenac and ibuprofen), with each of the two groups consisting of about 4000 arthritic patients, 72 percent of whom suffer from osteoarthritis (OA) (33). CLASS patients were additionally permitted to take low-dose aspirin as needed. There was no significant difference in the primary end point, the incidence of complicated ulcers (i.e., ulcers requiring hospitalization and resulting in significant morbidity and/or mortality) between the groups (0.7 percent for those patients receiving celecoxib vs 1.5 percent for those receiving either of the other two NSAIDs; P = 0.09), but the incidence of ulcer complications plus symptomatic ulcers was significantly lower for patients receiving celecoxib as opposed to ibuprofen or diclofenac (2 percent vs 3.5 percent; P = 0.02).
Thus, current data from two prospective studies generally support the COX-2 hypothesis. In practical terms, switching from a conventional NSAID to a COX-2 inhibitor can be estimated to halve the risk of PUB. In order to prevent a single incidence of a complicated ulcer, on statistical reasoning, only 500 low-risk or 40 high-risk patients would have to be switched to a coxib (34). Still, clinicians will have to consider those risk factors, such as age and prior history of ulcers or GI bleeding, that can markedly influence the occurrence of NSAID-induced PUB. Both of the studies discussed above excluded patients with prior symptomatic or recognized GI ulceration, where the risk of complicated ulcers caused by NSAIDs is about 5 percent, in contrast to an incidence of 0.4 percent for RA patients with no risk factors (35).
We still have much to learn about the precise roles of the two COX isoforms within the clinical setting of an established ulcer. Both COX-2 and COX-1 have been detected in apparently normal GI epithelium, raising the possibility that both may contribute to the generation of cytoprotective prostaglandins (36). GI epithelial COX-2 is upregulated by a range of insults, including infection with Helicobacter pylori (37, 38), and COX-2 expression in humans is increased at the margin of healing ulcers. COX-2 inhibitors also impair ulcer healing in mice (39), perhaps reflecting an effect on angiogenesis and/or the restoration of epithelial integrity (39, 40). Indeed, despite the encouraging data that indicate the decrease in GI toxicity associated with the coxibs relative to the traditional NSAIDs, the broader potential risks of COX-2 inhibition within the GI tract are still poorly understood. COX-2 products influence the intestinal response to antigens, and COX-2 inhibitors impair the tolerance of dietary antigens in mice (41) and worsen experimental colitis in rats (42). In fact, deletion of either mouse COX isoform exacerbates dextran-induced colon inflammation, and the inhibition of COX-2 in COX-1 knockout mice further exacerbates the response to dextran (43).
Implications of Selective COX Inhibitors in Cardiovascular Function
The COX hypothesis, strictly as we have presented it above, addresses the possible advantages of the coxibs with respect to the GI system. But it is clear that the relative levels of the COX-1 and COX-2 activities are important in other organ systems, and selective inhibition of COX-2 must thus be considered beyond the GI context. COX-1 is expressed constitutively in both endothelial and smooth muscle cells, and its products play a major role in homeostasis. Although the COX-2 isoform is more widely recognized for upregulation (in response to cytokines, growth factors, phorbol esters, LPS, and injury to vascular cells), evidence from foam cells and vascular smooth muscle cells in human atherosclerotic plaque indicates that COX-1 also can be upregulated (8).
A Proactive Role for COX-2 in Modulating Thrombosis
Among the important roles that COX-2 plays in terms of its induction in disease states is the production of prostacyclin associated with atherosclerotic platelet activation (44). Significantly, however, COX-2 may be active under normal physiological conditions as well. Indeed, physiological rates of shear upregulate COX-2 expression in endothelial cells in vitro (45), and selective COX-2 inhibitors depress urinary prostacyclin metabolites in healthy individuals (46-48). Prostacyclin—and COX-2—may thus represent part of a homeostatic defense mechanism that limits the consequences of platelet activation in vivo. Deletion of the prostacyclin receptor in mice leads to increased sensitivity to thrombotic stimuli, although it does not result in spontaneous thrombosis (49). COX-2 may be relevant to other aspects of cardiovascular biology as well. Suppression of COX-2–dependent prostacyclin formation, for example, increases the susceptibility of ventricular myocytes to oxidant injury in vitro (50). The effects of COX-2 inhibitors on prostacyclin formation may thus be important, although the potential risk of thrombosis is likely to be small due to several homeostatic defense mechanisms, such as those provided by nitric oxide and ectoADPase, that check platelet activation. Thrombosis has occurred acutely on celecoxib administration to four patients with lupus anticoagulant (51), but because such patients develop spontaneous thrombosis anyway, the relationship of these events to drug intake remains speculative.
COX-2 Inhibitors: Evidence for AdverseCardiovascular Events?
Major vascular events—primarily in the form of myocardial infarction—in the VIGOR study occurred more frequently with the administration of rofecoxib than with administration of naproxen (0.8 percent vs 0.4 percent; P<0.05). In the CLASS trial of celecoxib, on the other hand, where patients were permitted to take low-dose aspirin, there was no significant difference in the incidence of cardiovascular events between the main treatment groups (33). (The allowance of aspirin, however, may have mitigated any reduction in incidence of GI adverse events upon use of celecoxib as opposed to NSAID comparators.)
Furthermore, due to the low absolute number of cardiovascular events in the VIGOR study, their statistical significance remains uncertain. Indeed, RA patients such as those used in the study may have an increased risk of thrombotic events (52, 53). On the other hand, the absence of an increase in thrombotic events in the celecoxib study (CLASS) does not rule out the possible relevance of reduced prostacyclin production in response to coxib therapy. The prevalence of OA patients in CLASS, who do not show an increased propensity for thrombosis, along with the use of aspirin may have precluded observation of effects arising from diminished levels of prostacyclin. Alternatively, the comparators in VIGOR and CLASS evaluations may differ in their potential to offer cardioprotection. A “prothrombotic” drug, for example, could favor manifestation of the clinical effects of prostacyclin deficiency, but it is also conceivable that naproxen represents a cardioprotective comparator in the VIGOR scenario. Although there is no convincing evidence that NSAIDs, including naproxen, protect against cardiovascular events (54), naproxen, in fact, has an extended half-life and completely suppresses the capacity of platelets to make TxA2, a prostanoid that promotes vasoconstriction and platelet aggregation (55). Neither of these biologically plausible hypotheses—prostacyclin suppression by rofecoxib or cardioprotection by naproxen—has been established.
Pending further studies, what is a reasonable approach to the use of NSAIDs, both traditional and COX-2 selective? Both rofecoxib and celecoxib can be as effective against inflammation as the traditional NSAIDs at comparable concentrations, and the potential hazards due to suppression of prostacyclin production remain theoretical, although more study in this regard is clearly needed (46-48). Nevertheless, the administration of coxibs should be considered within the wider clinical context. Specifically, the cost effectiveness and considerable efficacy of aspirin in reducing both the primary and secondary incidence of vascular events has resulted in its wide use. Patients who have experienced a cardiovascular event in particular are advised to adhere to low-dose aspirin treatment. Thus, the widespread use of aspirin and other NSAIDs is an important factor in determining the appropriateness of additional, COX-2–selective therapy. If anti-arthritic medication is called for in patients already receiving low-dose aspirin, the CLASS analysis raises the possibility that the advantage of a coxib, in terms of an improved profile of GI effects, might be marginal. This issue needs to be addressed by appropriate clinical studies.
Conclusion
COX-2–selective inhibitors provide a unique example of accelerated drug discovery and development. In the past ten years or so, since the identification of the COX-2 isoform, targeted design of pharmaceutical agents has allowed remarkable advances in clinical applicability. COX-2 inhibitors cause significantly fewer serious GI adverse events than traditional NSAIDs. The prospect of additional uses for these drugs, such as in cancer prevention and delaying the progression of Alzheimer's disease, is of great interest. Even more selective COX-2 inhibitors are currently in clinical development. With further progress in understanding the biology of COX-2 and its clinical pharmacology, the therapeutic potential of coxibs is likely to expand.
Acknowledgments
The authors' work is supported by grants HL 62250, HL 57847 and M01 RR00040 from the National Institutes of Health.
- © American Society for Pharmacology and Experimental Theraputics 2001
References

Issam A. Mardini

Garret A. FitzGerald

