Neuropathic Pain: The Paradox of Dynorphin
- Departments of Pharmacology and Anesthesiology University of Arizona Health Sciences Center Tucson, Az 85724
- Address correspondence to FP. E-mail frankp{at}u.arizona.edu; fax 520-626-4182.
Abstract
One of the curious but common consequences of opioid administration in the clinical setting is the induction, at sites uninvolved in the original presentation of discomfort, of pain itself. The induction of pain is also a reliable, measurable phenomenon in animals receiving continuous delivery of opioid. Such pain induction is associated with the expression of spinal dynorphin, a finding that is especially intriguing in light of dynorphin's ability to recapitulate many of the characteristics of chronic, neuropathic pain when administered intrathecally (i.e., into the spine). The effective treatment of chronic pain syndromes—and of tolerance to antinociceptive therapies—may thus rest on an understanding of the biological roles of dynorphin in neurotransmission.

The dorsal horn cells seen above have been immunolabeled (green) with antiserum raised against prodynorphin, the precursor
peptide of the endogenous opioid. Unlike the other two recognized classes of endogenous opioids—the enkephalins and the endorphins—dynorphin
and its derivatives fail to function as effective analgesics upon spinal injection. Indeed, it appears that dynorphin has
important non-opioid functions that maintain neuropathic pain and that may thus represent viable targets of antinociceptive
therapy.
INTRODUCTION
Clinically, neuropathic pain is an intractable, chronic syndrome that may arise from injury to peripheral nerves and is associated with hyperalgesia, spontaneous pain, allodynia, repetitive discharge of nociceptors, and the expansion of receptive fields of nociceptive input (1, 2). Our understanding of the pathophysiology of this debilitating syndrome is very limited, and although several classes of compounds, including anticonvulsants, antiarrhythmics, and local anesthetics, show some efficacy against neuropathic pain (3), they are generally required at high dosages that elicit severe side effects. Furthermore, analgesics that are highly effective in the management of acute pain often provide only very short-term relief (if any at all) against neuropathic pain.
Clearly, an understanding of the mechanisms that underlie neuropathic pain is essential for the ultimate goal of devising effective treatments for this syndrome. A number of experimental models of neuropathic pain have been invaluable in evaluating the physiological and neurochemical changes that occur in the nervous system as a result of peripheral nerve injury and in assessing the relevance of these changes to the development of abnormal pain states. This review will focus on the role of the endogenous neuropeptide dynorphin in promoting neuropathic pain.
THE PARADOX OF DYNORPHIN
Although dynorphin is commonly characterized as an endogenous opioid peptide and was originally proposed as a ligand of the opioid kappa receptor (4), numerous studies have indicated that much of the pharmacology of this peptide is independent of interaction with opioid receptors (5). Unlike the other two classes of known endogenous opioid peptides, the enkephalins and endorphins, intrathecal administration of dynorphin—whose N-terminal sequence is identical to [Leu]enkephalin—does not elicit robust antinociception. Rather, dynorphin and those derivatives that lack the crucial N-terminal tyrosine residue necessary for interaction with opioid receptors (see Table 1⇓) produce motor impairment and paralysis. Intrathecally administered dynorphin and its non-opioid derivatives produce hindlimb paralysis (6, 7) and enhanced sensitivity to sensory stimuli; at paralytic doses, these peptides are neurotoxic, depleting sensory neurons, motor neurons, and interneurons in the spinal cord (8), and potentiating excitatory neurotransmitter release (9, 10). At non-paralytic doses, dynorphin elicits long-lasting hyperresponsiveness to innocuous mechanical and noxious thermal stimuli (11, 12). The essentially equivalent actions of dynorphin and its non-opioid derivatives strongly suggest that the effects of dynorphin are non-opioid. Increased responsiveness to innocuous and noxious sensory stimuli are reminiscent of signs of experimental and clinical neuropathic pain, suggesting that endogenous dynorphin is a key mediator of some aspects of neuropathic pain states.
Ki Values for Dynorphin Variants with Respect to Human Opioid Receptors
DYNORPHIN AND PAIN STATES
Nerve Injury
Interestingly, peripheral nerve injuries are accompanied by an increase in spinal dynorphin (13–16). Although expression levels of other spinal neuropeptides also change upon nerve injury, the upregulation of endogenous spinal dynorphin is of special interest in view of its non-opioid, hyperalgesic pharmacology.
The immunodetection of dynorphin normally predominates in spinal interneurons (17). Within five days subsequent to nerve injury, dynorphin concentrations are elevated in the dorsal horn (i.e., where afferent axons terminate), peaking at day ten and persisting past day twenty post injury (15); levels of mRNA for preprodynorphin correspondingly rise (14). Complete nerve transection or nerve crush injury, however, produces only minimal changes in preprodynorphin mRNA levels (14), suggesting that sustained afferent discharge is important for initiating the upregulation of spinal dynorphin. Our own studies have indicated significant increases in spinal dynorphin-like content in the ipsilateral dorsal quadrant of the spinal cord after injury to the L5 and L6 spinal nerves (18). The increase in dynorphin content, peaking approximately ten days after nerve injury and persisting for many weeks, is consistent with the duration of the experimental pain state. A bilateral increase in immunologically detected dynorphin content is also observed in a sciatic cryoneurolysis model of neuropathic pain (19). In polyarthritic rats, moreover, a twofold increase in dynorphin immunoreactivity can be detected in spinal cord perfusate collected from in vivo intrathecal catheters, along with a concomitant increase (four- to sevenfold) in spinal prodynorphin mRNA (20). Taken together, these studies suggest that the enhanced synthesis and release of spinal dynorphin after nerve injury may be common in chronic pain.
Although it has generally been concluded that elevated concentrations of spinal dynorphin function—in accordance with its classification as an endogenous opioid—to dampen chronic nociceptive input after nerve injury (14, 15), considerable evidence reveals that dynorphin is actually pronociceptive in chronic pain states. A pathogenic role for dynorphin is indicated, for instance, by the fact that spinal administration of anti-dynorphin antiserum reduces neurological impairment after nerve injury (21, 22). We have shown that the intrathecal injection of anti-dynorphin antiserum also blocks the increased sensitivity to noxious thermal and innocuous mechanical stimuli that commonly accompanies injury to spinal nerves, but that the antiserum does not alter normal sensory thresholds in non-injured animals (18, 23–25). These findings support the hypothesis that the upregulation of spinal dynorphin is pronociceptive and important in the maintenance of the experimental neuropathic pain state.
Prodynorphin Knockout Mice
The hypothesis that dynorphin is important in the maintenance of neuropathic pain states has been further tested using mice in which the prodynorphin gene is deleted (26). These mice generally show normal responses to innocuous and noxious stimuli, thereby suggesting that basal levels of spinal dynorphin do not substantially participate in sensory thresholds (24). They also develop, as do their wild-type litter mates, neuropathic pain within two to three days after nerve injury, manifesting the typical increase in sensitivity to innocuous mechanical and noxious thermal stimuli. Unlike their wild-type litter mates, however, the prodynorphin knockout mice fail to sustain the neuropathic pain state for extended periods. By ten days after injury, the knockout mice are no more sensitive than they were prior to injury. In contrast, wild-type animals display neuropathic pain past day fourteen (24). These data suggest that spinal dynorphin does not contribute to initial pain onset, but rather is critical for the maintenance of neuropathic pain. Correspondingly, early after nerve injury (ca two days), when experimental neuropathic pain is alleviated by spinal administration of the N-methyl-d-aspartate (NMDA) receptor antagonist MK-801, anti-dynorphin antiserum is ineffective. Significantly, anti-dynorphin antiserum blocks experimental neuropathic pain at ten days post injury, when spinal dynorphin levels are at their peak (18, 24). Collectively, these findings strongly support the upregulation of spinal dynorphin as a mechanism for the maintenance of the neuropathic pain state.
NMDA RECEPTOR ACTIVITY AND DYNORPHIN
Several investigators have demonstrated that dynorphin-induced neurological damage, such as hindlimb paralysis, loss of tail-flick reflex, and loss of neuronal cell bodies, can be prevented by antagonists and modulators of the NMDA receptor, which is known to be an important mediator of neuropathic pain states (27–32; 33, 34). Similarly, the long-lasting hyperresponsiveness to both innocuous and noxious stimuli that is elicited by intrathecal dynorphin is not blocked by the opioid receptor antagonist naloxone, but is prevented by pretreatment with the NMDA receptor antagonist MK-801 (11, 12). This finding suggests that the NMDA receptor is a likely mediator of the non-opioid actions of dynorphin.
The interaction of endogenous dynorphin and NMDA receptors in promoting neuropathic pain may be either direct or indirect. A direct interaction between dynorphin and NMDA receptors was first reported by Massardier and Hunt (35), who showed that dynorphin and its fragments could displace [3H]glutamate bound to rat brain membranes. The interaction between dynorphin and NMDA receptors has been subsequently substantiated by the use of [3H]MK-801 and other radioligands that are highly selective for NMDA receptors (36). Because MK-801 blocks NMDA receptors by binding selectively to open (i.e., active) channels (37), its displacement by dynorphin suggests that dynorphin either competes with MK-801 for binding to the open state or maintains NMDA receptors in the closed (i.e., inactive) state, which has low affinity for MK-801. Additional analysis supports the latter alternative: Dynorphin is a positive allosteric modulator of the binding of the competitive NMDA receptor antagonist CGP39,653 to the glutamate binding site (38). The apparent Kd of the dynorphin–NMDA receptor complex originally reported through such analysis was in the micromolar range. However, such indirect radioligand competition analysis may not truly reflect the affinity of dynorphin for the NMDA receptors if both dynorphin and the ligand occupying the NMDA binding site have modulatory effects on the conformation of the receptor complex. More recently, our laboratory has described the saturable, high-affinity (Kd=10 nM) binding of radiolabeled dynorphin to NMDA receptors in rat cortical brain membranes (39). Additionally, HEK293 cells transfected with NMDA receptor R1/R2A subunits similarly show high-affinity binding of dynorphin A(2–17) that is potentiated by antagonist binding at either the glutamate or the glycine binding sites (39), whereas agonists attenuate dynorphin A(2–17) binding (Figure 1⇓; J.Lai, Q.Tang, and F. Porreca, unpublished observation). Thus, both direct and indirect binding analyses indicate that dynorphin A associates more strongly with NMDA receptors that have high affinity for antagonists (i.e., that are in the closed channel state). Electrophysiological analysis has demonstrated that dynorphin directly blocks NMDA-activated currents in isolated trigeminal neurons (40). Dynorphin shortens the mean open time and decreases the probability of channel opening, but does not seem to affect single channel conductance. Dynorphin A(1–13) inhibits the NMDA-induced currents of recombinant, heteromeric NMDA receptors expressed in Xenopus oocytes, and this inhibition is dependent upon receptor subunit composition (41).
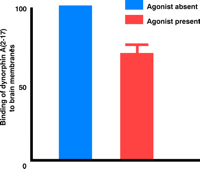
NMDA receptor agonists attenuate the specific binding of [3H]dynorphin A(2–17) to rat brain membranes. The specific binding of [3H]dynorphin A(2–17) was reduced by 30% in the collective presence of glutamate, glycine, and spermine.
a Units are arbitrary. Crude membranes (300 μg) from rat brain were incubated (5 mM Tris buffer, pH 7.5) with 1 nM [3H]dynorphin A(2–17) with or without agonists (10 μM glutamate, 10 μM glycine, and 10 μM spermidine) in a final volume of 0.5 mL for 2 h at 25°C. Non-specific binding was defined as the binding of the radiolabeled ligand in the presence of unlabeled 200 μM dynorphin A(2–17).
Although the sensitivity of NMDA-induced currents to dynorphin would seem to corroborate the selectivity of dynorphin for the closed state of NMDA receptor channels, inhibition by dynorphin runs contrary to the apparent excitatory and excitotoxic actions of dynorphin in vivo. Indeed, one study suggests that dynorphin can potentiate NMDA-induced currents under certain conditions (42). Specifically, dynorphin appears to potentiate recombinant NMDA receptors expressed in Xenopus oocytes at low glycine concentrations. Thus, although we cannot entirely rule out a direct, excitatory interaction between dynorphin and NMDA receptors, most evidence argues against direct interaction as a predominant mechanism for the excitatory actions of dynorphin, implying that non-opioid actions of dynorphin arise from interactions not involving NMDA receptors. Given the excitotoxicity of dynorphin, along with the neuropathic pain states associated with NMDA receptor activation, the physiological relevance of dynorphin as an inhibitory ligand of NMDA receptors is at present unclear. Indeed, substantial evidence suggests that dynorphin facilitates excitatory amino acid release, and because the binding of dynorphin to NMDA receptors is attenuated by NMDA agonists, dynorphin may not interact appreciably with NMDA receptors under neuronal hyperexcitable conditions.
DYNORPHIN AND NEUROTRANSMITTER RELEASE
The hypothesis that the biological roles of dynorphin extend beyond opioid and NMDA receptors is supported by the finding that the application of dynorphin to the rat hippocampus results in a localized, dose-dependent release of glutamate and aspartate (9). Thus, dynorphin may indirectly potentiate NMDA receptor activity by enhancing the release of excitatory neurotransmitters, thereby resulting, for example, in hindlimb paralysis and neuronal cell loss.
Recently, Hargreaves and colleagues have demonstrated that capsaicin, an activator of nociceptive C fibers, stimulates the release of the calcitonin gene related peptide (CGRP) from primary afferent terminals in spinal cord preparations, and that the release of this neuropeptide is promoted by dynorphin (43). This observation has been confirmed in our laboratory and is consistent with previous reports that dynorphin facilitates the capsaicin-evoked release of substance P from trigeminal nuclear tissue (44). It is feasible that dynorphin presynaptically promotes the release of these neuropeptides by facilitating the rise in intracellular calcium concentration that would accompany activation of NMDA receptors. Indeed, neurotransmitter release depends on the transient increase in calcium that facilitates the interaction of proteins that mediate the docking and fusion of synaptic vesicles to the plasma membrane at the axon terminal (45).
NEURONAL EXCITATORY ACTIONS OF DYNORPHIN
In order to examine whether dynorphin can promote an increase in intracellular calcium concentration ([Ca2+]i) in neuronal cells, we examined the acute effect of dynorphin on [Ca2+]i in individual cultured neurons isolated from neonatal rat cortex (Figure 2⇓). Specifically, we found that the non-opioid peptide dynorphin A(2-17) induces a time- and dose-dependent increase in [Ca2+]i (46). This effect was blocked neither by naloxone nor MK-801, even though the cells studied were responsive to NMDA. Thus, dynorphin appears to stimulate an acute increase in [Ca2+]i in cortical neurons, but does so in a manner that is independent of NMDA receptors. These findings implicate a novel site of action of dynorphin that may enhance neuronal excitability through an increase in intracellular calcium. Preliminary data from our laboratory also show that dynorphin similarly enhances [Ca2+]i in cultured dorsal root ganglion cells (primary afferent neurons), and potentiates the ability of capsaicin to evoke the release of CGRP from these cultures (J. Lai, unpublished observations). Thus, dynorphin may have a direct excitatory effect on neurotransmitter release from primary afferent nerves.
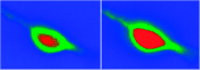
Dynorphin A(2–17) mediates an increase in intracellular calcium ion concentration in rat cortical neurons. The pseudocolor image shows the basal fluorescence of a typical neuron loaded with the calcium-sensitive fluorophore fluo-3 (left) and the same cell after the administration of 30 μM dynorphin A(2–17) to the bathing medium (right). Dynorphin A(2–17) induces a significant increase in the fluorescence of fluo-3 (expanded red area); the maximum fluorescence is reached after 1 min.
The novel mechanism of dynorphin-induced increase in [Ca2+]i is currently under investigation. Many potential calcium-dependent mechanisms could facilitate neurotransmitter release by dynorphin (Figure 3⇓). For example, the activation of protein kinase C enhances the release of CGRP from spinal cord slices (47) as well as from cultured sensory neurons (48). NMDA receptor–mediated currents are potentiated by protein kinase C and its activators (49, 50). In addition, many voltage-gated calcium and potassium conductances are modulated by calcium through calmodulin, which also regulates the activity of a number of kinases that have been implicated in the regulation of neurotransmitter release (51, 52) and in the phosphorylation of synaptic proteins (53).
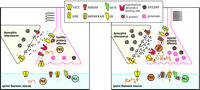
Possible mechanisms of dynorphin in maintaining neuropathic pain. Under normal conditions (left panel), dynorphin acts at opioid receptors (OR) to limit noxious input from a primary afferent: Specifically, dynorphin activates potassium efflux (through a G protein–coupled inwardly rectifying potassium channel; GIRK) and inhibits calcium influx (through a voltage-sensitive calcium channel; VSCC) in primary afferent and spinal thalamic neurons. Thus, excitatory neurotransmitter (glutamate) release is inhibited from the primary afferent terminal. After nerve injury (right panel), sustained input from the injured nerve drives the upregulation of spinal dynorphin through an unknown mechanism. The enhanced release of dynorphin stimulates the activity of molecules such as protein kinase C (PKC) and VSCC at the primary afferent to facilitate release of excitatory neurotransmitter and neuropeptides (e.g., substance P [SP] and CGRP). The binding site that mediates the effects of dynorphin is unknown. Enhanced neurotransmission potentiates the activity of postsynaptic glutamate receptors (AMPAR/KAIR) and NMDA receptors (NMDAR) by calcium/PKC dependent receptor phosphorylation. This is in part due to the activation of the substance P receptor (NK1R)/phospholipase C (PLC) pathway.
A recent study of cultured embryonic spinal cord neurons shows that dynorphin A(1-13) stimulates an acute, transient increase in [Ca2+]i (54). The excitatory effects of dynorphin are therefore apparent in both presynaptic (dorsal root ganglion) and postsynaptic (spinal cord) neurons. Prolonged exposure of these cells to dynorphin not only leads to an elevated basal level of [Ca2+]i, but also a significant loss of neurons; both of these effects are prevented by NMDA antagonists, suggesting that the neurotoxic effects of dynorphin are calcium dependent and mediated by NMDA receptors. These data support the notion that continuous exposure to dynorphin potentiates NMDA receptor activity, in accord with the neurotoxicity of intrathecal dynorphin to spinal cord neurons as mentioned above (8). The underlying assumption of Hauser et al. is that this effect is mediated by a direct binding of dynorphin to the NMDA receptors; however, the mechanisms by which dynorphin potentiates NMDA receptor activity remain equivocal due to the prolonged effect of dynorphin on [Ca2+]i through the other calcium dependent mechanisms as mentioned above. Furthermore, it remains to be established whether the acute effect of dynorphin on [Ca2+]i is mediated by NMDA receptors in spinal cord cells. Nevertheless, enhancement of spinal neuron excitability by dynorphin could contribute significantly to the hyperresponsiveness to sensory input observed after nerve injury.
CONCLUSIONS
Under normal physiological conditions, spinal dynorphin plays a minimal role in the regulation of sensory thresholds, a conclusion that is supported by prodynorphin knockout mice (24). Rather, the opioid action of dynorphin is to suppress noxious inputs, inasmuch as antiserum to dynorphin enhances formalin-induced flinching behaviors (55). However, under pathological conditions resulting from injury to peripheral nerves, the upregulation of spinal dynorphin accompanies the development of chronic pain states that can be blocked by anti-dynorphin antiserum, suggesting that elevated spinal dynorphin is required for maintaining neuropathic pain (Figure 3⇑). The mechanisms that mediate the upregulation of spinal dynorphin are unknown. Evidence from many laboratories consistently shows that neuronal excitation by dynorphin is mediated by an increase in intracellular calcium, which may act to enhance the release of transmitters from sensory primary afferent neurons or to potentiate NMDA receptor activity in spinal cord cells. The former possibility might represent a feed-forward mechanism by which spinal dynorphin could maintain an elevated level of sensory excitation, whereas the potentiation of NMDA receptors would greatly facilitate the responsiveness of spinal thalamic neurons to excitatory input. The pronociceptive actions of elevated spinal dynorphin suggest that the mechanisms by which dynorphin is upregulated, and by which dynorphin acts to promote excitation, are relevant to the development of therapeutic strategies to block chronic neuropathic pain.
- © American Society for Pharmacology and Experimental Theraputics 2001
References

(Clockwise from top): Michael H. Ossipov, Ph.D., is Assistant Professor of Pharmacology; Todd W. Vanderah, Ph.D., is Assistant Professor of Anesthesiology; Josephine Lai, Ph.D., is Associate Professor of Pharmacology; and Frank Porreca, Ph.D., is Professor of Pharmacology. Not shown: T. Philip Malan, Jr., M.D., Ph.D., is an anesthesiologist and Professor of Anesthesiology.

