Private Development Companies: Transforming Academic Research into New Treatment Options for Cancer
In 1971, President Nixon signed the National Cancer Act into law. Intended to eradicate cancer as a major cause of death, the Act functioned, in part, to make funds available for the identification of new molecular targets. Nearly forty years later, with expenditures of over $200 billion, the age-adjusted death rate for cancer in the US remains unchanged (1, 2). In contrast, over this same period of time, with expenditures approaching only half of those invested in cancer research, deaths from coronary disease declined from over 375 to under 150 per 100,00 individuals (3, 4). Early on, this disparity could have been attributed to our lack of an understanding of tumor cell biology. In the last fifteen years, however, our knowledge of both tumor cell biology and the human genome has grown exponentially, yet cancer deaths continue to surpass cardiovascular deaths (Figure 1). It is clear that we are not spending our money in a way that best leverages what we have funded and what we have learned. We urgently need new models for cancer drug development.
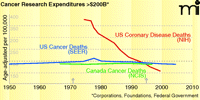
Age-adjusted death rates for cancer and coronary disease.
Private development companies could provide the means for meeting this need. Such companies would shepherd first-in-class molecules emerging from competitively funded academic oncology research centers into rigorous regulatory-directed development programs. These companies could “de-risk” experimental therapeutic innovations which, having no clearly defined pathway toward commercialization, often languish in the academic institutions where they are conceived. By providing translational pathways, private development companies would expedite the availability of these innovations to the marketplace by making them viable candidates for licensing to the pharmaceutical sector, which uniquely possesses the funding resources and expertise for clinical development and global marketing.
The current paucity of translational routes for antitumor drugs is a problem that can be linked to three key events that occurred in 1980. To understand the critical role that private development companies could play in solving this problem, it is worthwhile to understand its history, which involves a Supreme Court decision and two pieces of legislation. First, in Diamond v. Chakrabarty, the Supreme Court decided that genetically engineered microorganisms could be viewed as new compositions of matter. The litigants in this case were Al Chakrabarty, a General Electric microbiologist who developed a strain of Pseudomonas capable of using crude oil as a substrate, and Sydney Diamond, the US Commissioner of Patents, who in principle opposed patenting life. Chief Justice Burger, writing the majority opinion in a 5-4 decision, held that “any product of human ingenuity” was patentable. This landmark ruling enabled the nascent biotechnology industry to flourish. Although Genentech was founded four years earlier, in 1976, it would clearly rely on this decision. Also in 1980, Congress passed the Bayh-Dole Act (i.e., the University and Small Business Patent Procedure Act), which gave universities and research institutions the right to hold patents. The Act obligated patent-holding institutions to competitively license their intellectual property for maximum returns while giving preference to small businesses. These conditions essentially required the creation of technology transfer offices. Finally, Congress passed the Stevenson-Wydler Technology Innovation Act of 1980, which in turn required federal research institutions, along with universities, to attempt to competitively license their intellectual property.
As a consequence of the 1980 legislation, universities were placed upon a tripartite axis with pharmaceutical corporations and the newly emerging biotechnology industry (which they helped spawn and which grew in propinquity to them) as part of a plan to deliver the products of federally funded research back to the taxpayer and to improve the health of the American public (Figure 2). In this way, universities, for the first time, were actively encouraged to market their intellectual property. The legislation, however, failed to recognize the not-for-profit ethos in which academe is so deeply rooted. In fact, many universities had long-standing policies explicitly forbidding the marketing of biomedical research. As early as 1934, the President and Fellows of Harvard University had decided, “no patents primarily concerned with therapeutics or public health may be taken out by any member of the university.” Although these inveterate policies were rescinded in the 1970s, they continue to color the thinking at many universities. This, coupled with legal concerns associated with institutional tax-exempt status, still make it difficult for important drugs to reach the market.
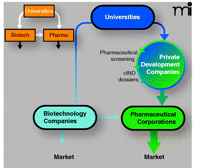
The University/Pharmaceutical/Biotechnology axis remodeled.
Upper inset. The familiar tripartite axis, representing a pathway for intellectual property to be developed into marketable therapeutics, resulted in 1980 from Diamond v Charkrabarty, the Bayh-Dole Act, and the Stevenson-Wydler Act (see text for details). White arrows represent the flow of intellectual property; black arrows represent a pathway to market. Main flowchart. In oncology, where the commercialization of effective drugs has been sluggish, private development companies are needed to transform academic research into new treatment options that reach the market. Private development companies would utilize experienced pharmaceutical review boards to select molecules emerging from competitively funded federal research programs and subject them to rigorous regulatory-directed development programs resulting in commercial Investigational New Drug (cIND) dossiers. These dossiers could be licensed to pharmaceutical clients, facilitating a route to market.
Nevertheless, research investments into the university/pharmaceutical/biotechnology axis have not been insignificant. In 2008, $29.8 billion of NIH funds, nearly $5 billion explicitly earmarked for cancer, flowed into universities and research institutions (4). Venture capital investments specifically targeted to biotechnology were slightly greater than $6 billion, more than double the amount invested across all sectors in 1990, and more than ten times the amount invested in 1980 (5, 6). Corporate research and development expenses at pharmaceutical corporations exceeded $50 billion, ten percent of which was spent extramurally (7).
Given the large sums of money that have been poured into drug development along the university-pharmaceutical-bio-technological axis, one must wonder what the return on this investment has been. If we focus on cancer, we find that six antitumor drugs accounted for one-third of the $66 billion in revenues posted by the twenty-five top-selling biotechnology drugs of 2008. Genentech developed the top three revenue producers worldwide: Rituxan, approved for non-Hodgkin’s lymphoma, grossed $5.1 billion; Avastin, approved for colorectal and non-small cell lung cancer, grossed $4.5 billion; and Herceptin, approved for first-line treatment of metastatic breast cancer, grossed $2.6 billion. Novartis’s Gleevac, for chronic myelogenous leukemia, grossed $3.7 billion worldwide, and Taxotere, a Sanofi-Aventis drug approved for second-line treatment of metastatic breast cancer, yielded $2.6 billion. Finally, Erbitux, approved for colorectal cancer, grossed $1.5 billion for ImClone. All in all, twenty-three cancer drugs had worldwide revenues of greater than $1 billion in 2008, and total worldwide revenues for antitumor drugs exceeded $40 billion (8).
Although the drugs we are developing for cancer produce significant revenue, their efficacies leave much to be desired. With the exception of Rituxan, which produced a five-year survival in lymphoma patients of over 50%, and Gleevac, which resulted in a one-year survival of 97% both invented by company scientists (9,10) the benefits these drugs provided, according to FDA-approved labeling at launch, have been less than stellar. Avastin extended survival by 4.7 months, whereas patients treated with Herceptin lived with no overall increase in tumor burden for an additional 2.7 months. Patients treated with Taxotere lived 2.7 months longer, whereas Erbitux gave patients the benefit of living only 6 weeks longer with no overall increase in tumor burden (11). In general, the rate of cancer drug approval in the US from 1987 to 2008 (about 3.5 drugs per annum) suggests that most cancer drugs that enter development do not work very well or do not work at all (12). And this must change.
This leads us to ask what our prospects might be for new effective antitumor agents. Data collected from some 160 universities and research institutions over ten years by the Association of University Technology Managers clearly suggests that, at present, we cannot rely on discoveries made in universities (13). Alarmingly, only one half of one percent of university patents licensed to industry produced income (both in fees and royalties) of greater than $1 million. With research expenditures reported at $200 billion and 25,000 patents licensed over the survey period, it now costs about $1.6 billion to produce a $1 million license. For large institutions (i.e., those spending over $250 million per annum on research), the median return on investment in 2007 was a paltry 1.5%, although the maximum return was 265.6% [the latter reflecting $791 million collected by New York University, largely associated with Remicade royalties (14)]. Despite the fact that the survey was not limited to biothera-peutics, to say we are inefficient in mining discoveries made in publicly funded research institutions, or that there is room for improvement, nevertheless, understates the dilemma.
The problem remains that universities and research institutions are not-for-profit organizations. As such, they do not warrant their patents, represent that these patents are not subject to competing blocking patents, nor guarantee the technical validity of their science. These institutions therefore license their intellectual property at a deep discount, to pharmaceutical clients who purchase these licenses defensively and, wary of the development risk, often do not exploit them. Thus, if inventions do not languish in universities, they languish in pharmaceutical corporations.
If we cannot rely on discoveries made in universities to populate a cancer discovery pipeline, what about privately funded premarket biotechnology companies? These companies have typically spun off from academic centers, taking with them, through technology transfer agreements, both key inventors and intellectual property. With regard to cancer therapeutics, such companies tend to position themselves for acquisition following proof-of-concept phase 2 trials. But it is difficult to see how this model can remain profitable. The issue is clinical development risk. Clinical development risk increases dramatically if clinical development is underfunded, as it almost always is at these small companies, reliant as they are on limited private equity funds. They typically can afford only one phase 2 trial per disease indication to create value.
The difficulty in cancer, in contrast to cardiovascular diseases, is that there are no biomarkers to allow companies to rationally pick dosing schedules, let alone determine activity after phase 1 data (15). This is likely why we have made such strides in treating coronary disease but not cancer. The clinical trials are so much easier. So for small companies focused on cancer, to expect real value accretion after limited phase 2 data is beyond wishful thinking. And this is where the bio-technology model fails in oncology.
Premarket biotechnology companies that undertake to develop new treatments for cancer, however, continue to be funded by a sequacious investment strategy that forces them to live or die on positive outcomes in one tumor type. Because it typically takes $100 million and eight to ten years to get to a point where success can be evaluated; because there is high clinical development risk; and because valuations must be extremely high in order to achieve a significant return on investment at acquisition: many investors are now starting to adopt an ABC policy (Anything But Cancer). This limits development opportunities for new drugs.
Private development companies offer the opportunity to address this dilemma by reorganizing the university/pharmaceutical/biotechnology axis (Figure 2). These companies would have access to a constellation of concepts already crystallized through federal funds, and could foster the development of many of the thousands of university patents issued annually [almost 4000 in 2007 (16)]. Alongside professional review boards and experienced oncology development teams, these private companies would work to find first-in-class molecules against novel targets, evaluate their patentability and existence of blocking patents, assess their pre-clinical efficacy, and determine their scalability and stability. Selected molecules would undergo intense regulatory-directed development studies which, if accomplished correctly, would allow the development company to compile commercial Investigational New Drug (cIND) dossiers. These dossiers could be competitively marketed to pharmaceutical clients (Figure 2).
In concept, a cIND dossier would be a ready-to-file document, comprising the major components of an IND following the common technical document format recognized by licensees and regulatory agencies worldwide. The cIND would homologate the pharmaceutical and regulatory development risks that often kill drug candidates between discovery and the onset of clinical trials. Moreover, a cIND could be warranted by the development company and thus of be of greater value than a university license.
Although they would compete with biotechnology companies for pharmaceutical clients, private development companies would structure themselves to license cIND dossiers rather than position themselves for acquisition. These companies would diversify risk by choosing from a number of first-in-class drug candidates, against novel targets, that are currently being produced in competitively funded federal research programs. With low internal costs a private development company could outsource most of its work each compound would require only a few million dollars in external costs over three years to reach a cIND endpoint that would generate license fees, and over time, milestone payments and royalties. Moreover, private development companies could become self-sustaining in a short time, depending on the value of the cINDs they produce.
One way to assess how a cIND might be valued in oncology is to assess how industry values early-stage data involving novel targets. Over the last year, deals made between bio-technology firms and pharmaceutical corporations involving inhibitors of novel targets with only incomplete phase 1 data garnered, according to company announcements, from $0.5 billion to just over $1.0 billion, excluding royalties. Examples of such deals include: i) an agreement between Bristol Myers Squibb and PDL BioPharma to develop a monoclonal antibody inhibitor of CS-1, a novel cell surface glycoprotein (upfront payment: $30 million; total deal value:$1.2 billion); ii) an agreement between Merck Serono and Lpath to develop a monoclonal antibody inhibitor of Sphingosine-1-phosphate (upfront payment: $23 million; total deal value: $0.5 billion); iii) an agreement between Sanofi-Aventis and Merrimack to develop a monoclonal antibody inhibitor of ErbB, a novel epidermal growth factor receptor (upfront payment: $60 million; total deal value: $0.5 billion); iv) an agreement between Onyx and S*Bio to develop a small molecule inhibitor of JAK2, a Janus kinase (upfront payment: $25 million; total deal value $0.6 billion); and v) an agreement between Sanofi-Aventis and Exelixis to develop a small-molecule inhibitor of PI3K, a phosphatidyl inositol 3 kinase (upfront payment:$140 million; total deal value $1.2 billion). Clearly, there is a perceived need and an appetite for early-stage data involving inhibitors of novel tumor targets. And without doubt, the gross margins of these deals would have been far greater had they involved private development companies operating with far less infrastructure compared to biotechnology companies.
To become a reality and to address the dearth of new treatment options for cancer, the need for private development companies must be understood by those who craft policy in the federal granting system, universities, and the private equity community. For too long it has been underappreciated that universities cannot efficiently license their patents to industry and that pre-market biotechnology companies cannot undertake the high commercial risk of developing new cancer therapies. Thus, the university/pharmaceutical/biotechnology tripartite axis dating from the 1980s, although formed with good intent, must be remodeled. Private development companies could re-invent a more productive architecture. By operating outside of universities, but in consort with them, they could add significant value to existing federally funded intellectual property and define a clear path to market for early-stage concepts. These companies would require relatively little cash, but their impact on cancer and other inadequately treated diseases could be large.
Acknowledgment
The author is grateful to Christine Swenson, PhD, for her insightful contributions to this work.
- Copyright © 2010

Andrew S. Janoff, PhD, was Chief Executive Officer of Celator Pharmaceuticals and its predecessor company, Celator Technologies, from 2002 to 2008. Previously he was a Vice President of Research and Development at Elan Corporation and, prior to that, Vice President and a Scientific Founder of The Liposome Company, Inc. He has managed numerous research and development programs from proof-of-concept and early stage drug discovery through IND and NDA filings. Email andrew.janoff{at}gmail.com.

