Networking with AKAPs
Context-dependent Regulation of Anchored Enzymes
Abstract
A-Kinase Anchoring Proteins (AKAPs) orchestrate and synchronize cellular events by tethering the cAMP-dependent protein kinase (PKA) and other signaling enzymes to organelles and membranes. The control of kinases and phosphatases that are held in proximity to activators, effectors, and substrates favors the rapid dissemination of information from one cellular location to the next. This article charts the inception of the PKA-anchoring hypothesis, the characterization of AKAPs and their nomenclature, and the physiological roles of context-specific AKAP signaling complexes.
Introduction
In the 1950s, Earl Sutherland defined how hormones stimulate the production of the second messenger 3′-5′-cyclic adenosine monophosphate (cAMP) by adenylyl cyclase (AC) (1). Today, the list of hormones, neurotransmitters, and effectors that work through cAMP is expansive. So why do organisms utilize such a diversity of ligands that all stimulate production of the same second messenger? Although it is generally accepted that tissue- and cell-specific expression of cAMP signaling components [such as kinases, guanine nucleotide exchange factors (GEFs), cyclic nucleotide gated ion channels, and phosphodiesterases] create an element of selectivity in these second messenger responses [reviewed in (2–5)], it is evident that other factors organize these enzymes to favor the catalysis of particular intracellular responses. This review works from the premise that this signal specificity is achieved through the local activation of signaling enzymes that are anchored to subcellular organelles and membranes. In particular, we highlight the A-kinase anchoring proteins (AKAPs), a family of functionally related intracellular proteins that coordinate and control cAMP-responsive events and a range of other second messenger mediated cellular processes.
Protein Kinase A
A recognized function of AKAPs is to tether the cAMP-dependent protein kinase (PKA or A-kinase) holoenzyme. The PKA holoen-zyme is a heterotetramer consisting of two regulatory (R) subunits that maintain two catalytic (C) subunits in an inhibited state (6). This enzyme is the principal intracellular receptor of cAMP (7). When cAMP levels are low, the PKA holoenzyme is dormant; however, when cAMP levels are elevated, two molecules bind to each R subunit, thereby releasing the active C subunits. The PKA C subunits phosphorylate serine–threonine residues on target substrates, typically within the sequence context of -R-R-X-S/T-X (8). The prominence of PKA as a principal mediator of cAMP responsive events is reflected by the prevalence of multiple R and C subunit genes in mammalian genomes. Two C subunit genes encode the ubiquitously expressed Cα and Cβ isoforms whereas a third isoform, Cγ, is found in primate testis. The four R subunit genes are subdivided into two classes: RI (RIα and RIβ) and RII (RIIα and RIIβ). Although all R subunits share 75% identity in their cAMP binding pockets, the RI and RII classes differ in their sensitivity to cAMP, phosphorylation patterns, and subcellular location (9, 10).
Discovery and Identification of AKAPS
It is now widely accepted that subcellular targeting of R subunits occurs through interaction with AKAPs. This growing family of forty-three functionally related genes is defined on the basis of their ability to interact with the R subunits of PKA. The first anchoring proteins were detected as protein contaminants that co-purified with R subunits on cAMP-agarose affinity columns (11). Later, the use of far-western blotting protocols that utilized RII as the probe, expression-cloning strategies, and yeast two-hybrid analyses uncovered many more members of the AKAP family (12–14). As the number of AKAPs increased, a rather haphazard nomenclature evolved. AKAPs are often named on the basis of their apparent molecular weight when detected in the far-western assay (generally referred to as the “RII overlay”). Slight differences in the electrophoretic mobility of AKAP orthologs sometimes led to a disparity in naming of the same anchoring protein in different species (15). Consequently, the Human Genome Organization (HUGO) proposed an alternative nomenclature that categorized anchoring proteins according to their gene name. For example, bovine AKAP75, human AKAP79, and murine AKAP150 all refer to the products of the AKAP5 gene (15–17). However, there are significant limitations to this logical AKAP gene nomenclature, as only fourteen of the forty-three currently known AKAP protein family members are designated in this manner. Furthermore, a recurring feature of AKAPs is intricate alternative splicing patterns, resulting in multiple isoforms of the same gene. The AKAP9 gene, for example, has six known splice variants including yotiao, AKAP350, AKAP450, and centrosome- and golgi-localized PKN-associated protein (CG-NAP) (18–23). Thus, a complex and often cumbersome nomenclature has arisen where the protein products of a single AKAP gene often have multiple designations. Table 1 lists the various names and subcellular locations of selected AKAP isoforms and orthologs and includes the HUGO gene nomenclature.
Nomenclature and Subcellular Localization for AKAPs, Splice Variants, Isoforms, Orthologs, and Associating Molecules
Structure and Function of AKAP Binding to PKA
Despite idiosyncrasies in the nomenclature, the singular, defining feature of an AKAP remains the ability to anchor the PKA holoenzyme. This protein-protein interaction involves an α-helical region on each anchoring protein that binds tightly to a docking/ dimerization (D/D) domain formed by the first forty-five amino acids of each RII monomer. Structural analyses reveals that the D/D domain folds into an antiparallel X type four-helix bundle that creates a docking site for the AKAP followed by twenty amino acids which are responsible for RII homodimerization (24–28) (Figure 1A, B). Most AKAPs exhibit high affinity for the RII subunit of PKA; however, the dual-specificity AKAPs (e.g., ezrin, D-AKAP1, D-AKAP2, merlin, and AKAP220), are able to interact with RI dimers, although typically with 10-100–fold lower affinity (29). The apparent lower affinity of RI for AKAPs can be attributed to structural differences in the N-terminal D/D domain of RI, leading to a faster dissociation rate for RI than for RII from AKAP’s amphipathic helix in vitro; however, the dynamics of these protein interactions in vivo is less well-studied.
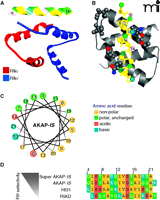
Structural aspects of AKAP interactions with the regulatory subunits of PKA. A. Structure of AKAP-IS in complex with the RII X-type 4 helix bundle. The AKAP-IS helix (top) interacts with the dimerization/docking (D/D) domain of RII through interactions within the core hydrophobic interface (yellow), and polar contacts that form hydrogen bonds with RII. B. Detailed top-view of the protein-protein interactions between AKAP-IS and RII. The residues important for AKAP-RII interactions (hydrophobic and H bonding) are numbered, with corresponding interaction sites in RII highlighted. C. Helical wheel rendering of the AKAP-IS amphipathic helix, showing a face of hydrophobic residues (yellow). D. Sequence alignment of the RII binding domain of AKAP peptides. The pdb accession number 2izx was used in A and B.
The reciprocal binding surface on the anchoring protein is an amphipathic helix (15, 24). The structural organization of this 14–18 amino-acid helix places hydrophobic residues on the interior face of the helix and hydrophilic residues on the exterior surface (Figure 1B, C). This creates a high-affinity binding surface that fits into a groove formed by the D/D domain of the R subunits. Any mutations that perturb the conformation of this helix have deleterious effects on PKA anchoring. Historically, helix perturbation (and the importance of individual resudes within the helix) has been demonstrated by substitution of various residues with proline, thereby ablating the high affinity RII interaction (15, 24). RI binding to dual-specificity AKAPs can be enhanced through an additional sequence outside of the amphipathic helix, termed the RI specifier region (RISR), originally identified in ezrin (30). To date, there are few reports of RI-specific AKAPs, including PAP7 (31), which may be a consequence of technical difficulties with the screening for RI interacting proteins or that the structure of RI has proven to be less accommodating to variations in the amphipathic helix than RII (32). However, there is reason to believe that a select few proteins may anchor PKA through alternative means. For example, the cytoplasmic tail of integrin α4, which is not predicted to form an amphipathic helix, binds to a C-terminal region of the type I regulatory subunit of PKA (33). Likewise, pericentrin tethers PKA to the centrosome through a 100 amino-acid domain in peri-centrin that interacts with a leucine rich region of RII (34).
A functional role for PKA anchoring is frequently inferred by delivery of anchoring inhibitor peptides. Peptides derived from the amphipathic helix of AKAP-Lbc, also known as human thyroid 31 (Ht31), were initially characterized for their ability to disrupt the PKA interaction with AKAPs (35, 36). Employing the optimal Ht31 peptide (amino acids 493–515), the function of AKAP-anchored PKA has been investigated using biochemical analyses, electrophysiology, and multiple cell-based methods to discern the role of AKAPs in cardiac excitation-contraction (EC) coupling, synaptic plasticity, reproduction, insulin secretion, and renal homeostasis (36–41). In some cases, cell-permeable stearated or myristoylated Ht31 peptides or plasmid-based transfection methods were used to overcome impermeability of cell membranes. Subsequent bioinformatic and peptide array studies led to the development of the AKAP–in silico (AKAP-IS) peptide, and a cell permeable TAT-AKAP-IS, which are selective for RII over RI (Figure 1D) (42, 43). Complementary studies led to the development of two RI-selective peptides; PV-38 and RI anchoring disruptor (RIAD), with RIAD having twentyfold higher potency than PV-38 and 1000-fold selectivity for RI over RII (32, 44, 45). Further honing of these reagents through high-resolution structural analyses has shown which residues in the amphipathic helix contact the R subunit dimer (Figure 1B) (46, 47). These studies have led to the development of the next generation of RI and RII selective peptide disruptors. For example, RIAD-RISR exhibits increased selectivity and affinity for RI than its predecessor RIAD (30). Likewise, SuperAKAP-IS has been reported to exhibit a fourfold greater selectivity for the displacement of anchored type II PKA than the original AKAP-IS (Figure 1D) (46). Although the selectivity and of these peptides and their further characterization was calculated in vitro, RIAD-RISR was recently utilized to implicate a role for RI-anchored AKAPs in T cell function, and AKAP-IS was used to implicate RII-anchored AKAPs in antigen presentation (44, 48). Future studies will likely elucidate the specific AKAPs involved in these immunological responses.
AKAPS Organize Multivalent Complexes
Another property of AKAPs is the ability to simultaneously associate with several binding partners (Table 1). This concept has been well established by numerous subcellular fractionation, yeast two-hybrid screening, and mass spectrometry approaches. The net result of these screens has been clear evidence that most, if not all, AKAPs have the capacity to interact with different combinations of activators, effectors, signal transduction and termination enzymes, and substrates (49, 50). This confers the ability of AKAP signaling complexes to facilitate rapid and efficient signal transmission in local environments. Some AKAPs are ubiquitously expressed, whereas others serve a specific function in specialized cell types. For example, in cardiomyocytes, numerous AKAPs occupy precise cellular locations to coordinate critical functions of the heart, including excitation-contraction coupling, oxygen homeostasis, energy expenditure, and transcriptional control (Figure 2A), AKAP-Lbc, for instance, is localized to the cytosol to coordinate multiple cardiac signaling events discussed in detail below (Figure 2B). This review will highlight progress in our understanding of the AKAP-Lbc and AKAP79/150 signaling complexes.
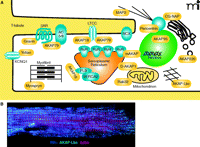
The coordination of PKA signaling events in a cardiac myocyte by localized scaffolding of AKAPs. A. In a cardiomyocyte, multiple AKAPs (yellow) coordinate physiologic and pathophysiologic signaling events, including excitation-contraction coupling, hypertrophic remodeling, gene transcription, and oxygen homeostasis. B. Adult mouse cardio-myocytes were fixed and incubated with antibodies against RIIα (blue) and AKAP-Lbc (green), and actin was visualized by phalloidin staining (red). Cells were then imaged using immunofluorescence microscopy. RyR, ryanodine receptor; βAR, β-adrenergic receptor; NCX, sodium-calcium exchangr ; CG-NAP, centrosome- and golgi-localized PKN-associated protein; MAP2, mitochondrial-associated protein 2 ; LTCC, L-type calcium channel; AC5/6, adenylyl cyclase 5 or 6; PLB, phospholamban; SERCA2, Sarcoplasmic-endoplasmic calcium pump 2.
The ability to integrate spatially constituents of different signaling pathways is clearly illustrated by AKAP-Lbc. This multipur-pose signaling molecule not only anchors several protein kinases but also functions as a guanine nucleotide exchange factor (GEF) for the small-molecular-weight GTPase Rho (51–53). As a result, AKAP-Lbc has been implicated in the control of diverse signaling events that range from the synchronization of transcriptional reprogramming events in the heart to the control of cell shape and motility in transformed cells. The PKA anchoring region of this molecule, which contains the sequence of the Ht31 peptide, has been exhaustively used to define the biochemical parameters of PKA-AKAP interactions (35). However, subsequent analyses have shown that this anchoring protein also acts as a scaffold for PKCs and PKDs (51). We have shown that the AKAP-Lbc associated pools of atypical PKCη and PKA act sequentially to generate an activation cascade for PKDs (Figure 3). This two-step process is initiated by PKC phosphorylation of sites in the catalytic core of PKD, whereas PKA phosphorylation of AKAP-Lbc at serine 2737 releases active PKD from the scaffold (51). This signaling complex functions in the heart where AKAP-Lbc integrates signals from α1-adrenergic and endothelin receptors to mobilize the PKD activation cascade (54, 55). The analysis of tissue samples from diseased hearts revealed that the expression of AKAP-Lbc is increased in response to hypertrophic stimuli (54, 55). Increased amounts of this anchoring protein augment signaling through a PKD-mediated transcriptional activation pathway that elicits a hypertrophic gene reprogramming phenomenon resulting in the reversion of cardiomyocytes to the developmental state, known as the fetal gene response. Live-cell imaging of cardiomyocytes reveal that the rate of nuclear export of the DNA effector protein HDAC5 was doubled in the presence of AKAP-Lbc (Figure 3) (55). Consequently, gene reprogramming via the myocyte-specific enhancer-binding factor (MEF-2) pathway was accelerated. Recently these findings have been corroborated and extended by studies in genetically modified mice that uncover a critical requirement for the AKAP-Lbc/AKAP13 gene in the developing heart (56).
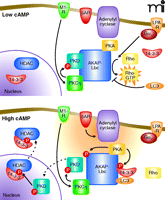
AKAP-Lbc integrates multiple signaling pathways. GPCRs such as M1 muscarinic receptor (M1-R) initiate a signaling cascade involving PKC-mediated activation of PKD and phosphorylation of HDAC in the nucleus. In another pathway, Gα12-coupled receptors such as the lysophospholipid receptor (LPA-R) stimulate the intrinsic Rho-GEF activity of AKAP-Lbc. When cAMP concentrations are low (top panel), PKD activity is relatively low and Rho-GEF activity relatively high. Gαs-coupled receptors such as βAR stimulate production of cAMP (orange cloud) by adenylyl cyclases (bottom panel), leading to the activation of PKA. Phosphorylation of AKAP-Lbc by PKA accentuates PKD signaling and curtails Rho-GEF activity. For simplicity, both panels show conditions where Gαq/11 and Gα12 pathways are active. HDAC, histone deacetylase; LPA-R, Lysophosphatidic acid receptor; βAR, beta-adrenergic receptor; LC3, microtubule associated protein light chain 3.
AKAP-Lbc also contains tandem Dbl homology–pleckstrin homology (DH-PH) domains that function as a Rho-GEF. The guanine nucleotide exchange activity residing in AKAP-Lbc is stimulated by the heterotrimeric G protein Gα12. Cell-based studies have shown that overexpression of AKAP-Lbc favors remodeling of the actin cytoskeleton to increase stress-fiber formation (53). Suppression of Rho activation, however, involves two AKAP-Lbc binding partners (57–59). First, AKAP-Lbc is phosphorylated by PKA at serine 1565 to promote oligomerization and the binding of 14-3-3 proteins (Figure 3) (60). This prevents the recruitment and GTP loading of Rho (57, 58). Second, binding of the ubiquitin-like protein LC3 prevents Rho interaction with AKAP-Lbc (59). It has yet to be established whether 14-3-3 and LC3 act synergistically to modulate GEF activity or whether both proteins simultaneously associate with this anchoring protein. Nevertheless, these findings illustrate how different combinations of AKAP-Lbc binding partners can influence diverse cellular processes and precisely control of intrinsic enzyme activity.
The capacity to interact with multiple binding partners to form customized signaling units may be best exemplified by AKAP79/150. This anchoring protein, initially discovered as an RII binding protein, was later shown to anchor the calcium/phospholipid-dependent kinase (PKC) and the calcium-calmodulin–dependent phosphatase (PP2B, also known as calcineurin) (15, 16, 61, 62). AKAP79/150 signaling complexes reside on the inner face of the plasma membrane in neurons, cardiomyocytes, and a limited number of other cell types (15). At this location, the anchored enzymes are optimally positioned to respond to the generation of intracellular second messengers such as cAMP, calcium, and phospholipid (63). Molecular and cellular approaches have further demonstrated that AKAP79/150 directs its cohort of anchored enzymes toward selected transmembrane proteins to facilitate their efficient regulation (Figure 4).

AKAP79/150 forms customized signaling complexes. Different combinations of enzymes and substrates are anchored by AKAP79/150. A. PKA stimulates calcium channel (Cav1.2) currents while also negatively regulating the action of adrenergic receptors and adenylyl cyclase, which are responsible for PKA activation. B. Signaling through the M1 muscarinic receptor activates PKC which phosphorylates and terminates M-current through the potassium channel KCNQ2. C. SAP97 recruits AKAP79/150 and its anchored enzymes PP2B and PKA, whose opposing actions regulate amino-3-hydroxy-5-methylisoxazole-4-propionate (AMPA) current through GluR1.
One role for these signaling complexes is the regulation of cAMP production (64, 65). AKAP79/150 is one of the anchoring proteins that directs PKA toward β-adrenergic receptors and plays a role in their phosphorylation-dependent downregulation (Figure 4A) (64–66). The same AKAP signaling complex also coordinates PKC-mediated phosphorylation events that influence the onset of angiotensin II-induced hypertension by increasing the likelihood of persistent CaV1.2 channel activity (67, 68). In other contexts, AKAP79/150 physically associates with the adenylyl cyclase 5 isoform, favoring phosphorylation of the enzyme to terminate cAMP synthesis. This finding supports real-time cellular imaging experiments showing that PKA anchoring to adenylyl cyclase 5 ensures rapid termination of cAMP signaling upon activation of the kinase (64, 69). This protein configuration permits the formation of a negative-feedback loop that temporally regulates cAMP production in the heart and endothelial cells (70). This important signal termination process can be achieved by altering the action of two AKAP79/150-associated proteins. PKA phosphorylation of β-adrenergic receptors leads to the desensitization and down-regulation of these receptors and shuts off adenylyl cyclase activity to cease the production of cAMP.
Another fascinating aspect of AKAP79/150 is that this anchoring protein can also couple its anchored enzymes to selected substrates. Functional studies in multiple cell types have shown that different AKAP79/150 complexes regulate the activity of ion channels. For example, direct association of AKAP79/150 with ion channels, such as the L-type calcium channels or M-type K+ channels, can regulate the activity of these channels (Figure 4A, B) (71–73). However, in other contexts, intermediary scaffolding proteins such as PSD-95 and SAP-97 link AKAP79/150 to synaptic glutamate receptor ion channels (Figure 4C) (71–76). The molecular details of these protein-protein interactions have recently been reviewed elsewhere (49, 50).
These findings emphasize the flexible design of the AKAP79/150 scaffold that allows it to not only to control cAMP generation through the modulation of G protein–coupled receptor and adenylyl cyclase activity, but also to facilitate the phosphoryla-tion dependent modulation of ion channels. Thus, the next level of study is to assess the individual contributions of each anchored enzyme in downstream signaling events. This exploration is partially illustrated by the opposing roles for PKA and PP2B on AMPA receptor currents, a result that was obtained by systematic introduction of mutant forms of AKAP150 that could not anchor PKA, PKC, or PP2B in neurons where the expression of the endogenous anchoring protein was silenced (73, 74).
The contribution of individual AKAP79/150 signaling complexes may also be assessed in AKAP150 knock-out mice. These animals exhibit reduced cerebellum-mediated coordination of motor control and altered long-term depression (LTD), further emphasizing the role of AKAP150 in anchoring PKA and additional enzymes in neurons (77). To dissect the role of AKAP150-anchored PKA, a line of mice was generated where AKAP150 was mutated to preclude PKA binding (78). This protein was designated Δ36, as a stop codon was introduced to prevent translation of the last thirty-six amino acids, which encompass the amphipathic helix. The resulting mouse displayed perturbations in multiple neuronal functions, including reduced long-term potentiation (LTP) in the hippocampus, which is involved in learning and memory. Ongoing studies that compare the phenotype of the AKAP150 knock-out to a range of AKAP150 knock-in mice that express forms of the anchoring protein that bring together selected combinations of enzyme binding partners are likely to shed light on the unique roles of anchored PKA, PKC, and PP2B in neurons and other tissues.
Measuring the Timing and Efficiency of Anchored Kinase Activity
Although AKAPs function to control spatially enzyme activity, they also contribute to the temporal resolution of signaling events. Live-cell imaging has proven to be the most useful technique to study this phenomena. Accordingly, a fluorescence resonance energy transfer (FRET)-based PKA activity reporter (AKAR) was developed and adapted to function like an AKAP (79, 80). The rate of AKAR activation is increased when PKA is anchored to the reporter, demonstrating the influence of kinase compartmentalization for optimal signal transduction. Subsequent studies have utilized a third generation AKAR (AKAR3) to visualize the activation of PKA in distinct subcellular compartments (81). Using wild type and AKAP79 mutants that cannot bind specific targeting enzymes, combined with electrophysiological measurements, AKAP79 was found to provide distinct subsets of enzyme complexes to regulate the M-type K+, AMPA, and GluRI channel activities (Figure 4) (73, 82–84).
Recently, AKARs and other fluorescent kinase reporters for PKC and Erk have been used to investigate how anchored kinases can influence the timing and efficiency of channel activity. More specifically, FRET kinase activity reporters were fused to AKAP79 to probe how the signaling complex functions to regulate the M-type KCNQ2 channel (84). Whereas PKA does not appear to be involved in this signaling process, PKC is known to phosphorylate serine 541 on the channel, leading to termination of M-current (Figure 4B) (83). Using the PKC FRET reporter C-kinase activity reporter (CKAR), simultaneous measurements of current and PKC activity were recorded upon stimulation with the muscarinic agonist oxotremorine-M (Oxo-M). Oxo-M stimulated a rapid suppression of the KCNQ2 current with a t½ occurring 13.5 seconds post stimulation, whereas the increase in CKAR activity had a t½ of 24.2 seconds post stimulation (84). To emulate anchoring of PKC, AKAP79 was fused to CKAR and the experiment was repeated. Strikingly, under these conditions there was no delay in PKC activity as compared to the timing of channel current (84). These results highlight how AKAP79 can optimize the transmission of the muscarinic signals to generate rapid and transient KCNQ2 channel activity.
AKAP79 Modifies the Pharmacodynamic Properties of Anchored Kinases
Another emerging principle is the ability of AKAP79 to modify the catalytic activity and solvent accessibility of its anchored enzymes. AKAP79/150 inhibits PP2B-mediated dephosphorylation of the nuclear factor of activated T cells (NFAT) transcription factor (61, 85). However, more recent pharmacological studies visualized anchored PKC-mediated phosphorylation of CKAR concurrently with electrophysiological changes in KCNQ2 channels to show that the AKAP79-bound PKC is resistant to the small-molecule competitive ATP analog inhibitors staurosporine and bisindolylma-leimide. Using the same system, it was shown that anchored PKC responds to calphostin C, another small-molecule inhibitor that targets a different region of PKC (84). The ramifications of this discovery may reach well beyond the AKAP field as it implies that signaling complexes may acquire reduced sensitivities to small-molecule inhibitors. As a result, these protein-protein interactions may create pockets of active kinase in situ and influence the pharmacological profile of certain kinase inhibitor drugs. This could have important consequences for drug discovery and research projects predicated on the selectivity of protein kinase inhibitors.
Conclusions
Nearly two decades have passed since the first discovery of AKAPs. In that time we have come to appreciate that AKAPs are found in every tissue, orchestrate various signaling mechanisms at all stages of life, and assemble a variety of signaling enzymes into localized signaling units. With so much diversity in localization mechanisms, a few general principles can still be drawn. The first seems obvious but deserves mention: AKAPs are almost invariably associated with membranes or other particulate cell fractions. Second, very few AKAPs are found inside the nucleus, which is not surprising given that PKA R subunits are also excluded. Third, precise positioning of AKAPs requires targeting to specific sub-cellular locations through protein-lipid interactions followed by protein-protein interactions within those regions. Fourth, multiple AKAPs are expected to occupy the same microdomain and even to simultaneously reside in the same signaling complex (Figure 2B).
Although the AKAP field remains very much in the discovery phase, and the identification of the components in each AKAP signaling complex remains a priority, several new concepts have appeared on the horizon. Can each of the known interacting partners of a particular AKAP bind simultaneously and how dynamic is the organization the complexes? Does one binding partner preferentially interact with the anchoring protein at the expense of another and what regulates that preference? The resolution of the 3-dimensional organization of these multivalent complexes will certainly yield valuable information regarding their functions. In addition, the further characterization of mutant AKAPs with specific domains deleted should reveal how the multiple signaling molecules cooperate through AKAP interactions. Collectively, these approaches should shed more light on the physiological roles of AKAP complexes in normal and pathological states.
Acknowledgments
The authors thank Matthew Gold, Catherine Pawson, and members of the Scott lab for critical evaluation of this article. JDS is supported in part by the National Institutes of Health National Institute of Diabetes and Digestive and Kidney Diseases [Grant P01DK54441] and the Leducq Transatlantic Network. EJW is supported by the National Institutes of Health National Heart, Lung, and Blood Institute [Grant DK54441].
- Copyright © 2010
References

John D. Scott, PhD, is the Edwin G. Krebs–Hilma Speights Professor in the Department of Pharmacology at the University of Washington and is a Howard Hughes Medical Institute Investigator. He is a Fellow of the Royal Society, London and the Royal Society of Edinburgh. He is interested in the specificity of signal transduction events that are controlled by anchoring proteins. Send correspondence to JDS: E-mail scottjdw{at}u.washington.edu; fax 206-616-3386.

Brian W. Jones, PhD, is a senior fellow working for Dr. Stanley McKnight and Dr. John Scott in the Department of Pharmacology at the University of Washington. He completed his doctoral work in Pharmacology at the University of Rochester, where he studied phosphorylation of G protein–coupled receptors. His current research focuses on the role of PKA anchoring by AKAPs in the heart and brain.
Emily J. Welch, PhD, joined John Scott’s laboratory in the Department of Pharmacology at the University of Washington in 2009, after receiving her PhD in Pharmacology from the University of Illinois at Chicago. While working toward her doctorate, she studied phospholipid signaling and metabolism. She currently investigates the roles of AKAP signaling complexes in cardiac physiology.

