Mechanism of Cancer Pain
- 1 Department of Oral and Maxillofacial Surgery, School of Dentistry, University of California San Francisco
- 2 Department of Diagnostic and Biological Sciences, School of Dentistry and Center for Pain Research, University of Minnesota, Minneapolis
- 3 Departments of Neuroscience, Pharmacology and Dermatology, Center for Pain Research and Medical School, University of Minnesota, Minneapolis
Abstract
Ongoing and breakthrough pain is a primary concern for the cancer patient. Although the etiology of cancer pain remains unclear, animal models of cancer pain have allowed investigators to unravel some of the cancer-induced neuropathologic processes that occur in the region of tumor growth and in the dorsal horn of the spinal cord. Within the cancer microenvironment, cancer and immune cells produce and secrete mediators that activate and sensitize primary afferent nociceptors. Pursuant to these peripheral changes, nociceptive secondary neurons in spinal cord exhibit increased spontaneous activity and enhanced responsiveness to three modes of noxious stimulation: heat, cold, and mechanical stimuli. As our understanding of the peripheral and central mechanisms that underlie cancer pain improves, targeted analgesics for the cancer patient will likely follow.
Introduction
Over half of all cancer patients will experience severe, uncontrollable pain during the course of their disease, and the management of pain is a primary challenge for the cancer patient and the treating oncologist (1). Although cancer pain is a complex pathologic process and a formidable clinical problem, significant headway has been made in understanding the basic neurologic mechanisms that are responsible for generating cancer pain. The symptoms experienced by the cancer patient are a consequence of cellular, tissue, and systemic changes that occur during proliferation, invasion, and metastasis. The responding immune system also has a clear role in cancer pain. The cancer cell produces mediators that affect other cells within the cancer microenvironment, such as immune cells. Nociception almost certainly involves dynamic interactions and crosstalk between the cancer and the primary afferent nociceptor. Therefore, it is difficult to isolate one cell and study it in isolation. The investigator must consider the activities of the cancer cell, the peripheral and central nervous system, and the immune system.
In the first part of this review, we discuss the relationship between the cancer cell and the primary afferent nociceptors within the cancer microenvironment. We focus on the mediators that are liberated by the cancer and sensitize the nociceptors; endogenous antinociceptive mechanisms in the setting of cancer are also reviewed. Other cells, particularly immune cells, near the cancer microenvironment will also be considered in the context of pain experienced by the patient and the level of nociception measured in the animal model.
The second half of this review discusses significant neurochemical changes in both the peripheral and central nervous system (CNS) in cancer pain. Electrophysiological and immunohistochemical studies have revealed signs of peripheral neuropathy as well as peripheral and central sensitization after tumor implantation in or around the limb bones of rodents. Tumor-related signs in rodent models mimic similar changes seen clinically, thus validating the models and shedding light on the mechanisms of cancer-evoked pain. Recent and thorough coverage of the broad range of animal models of cancer pain can be found elsewhere (2) and will be addressed here only briefly. Our focus will rather concern peripheral neuropathic changes and central sensitization that develop due to the influence of tumors on neurons.
Origins of Cancer Pain in the Microenvironment
The clinical presentation of cancer pain depends on three features: 1) the histologic type of the cancer; 2) the location of the primary neoplasm; and 3) location of metastases. A patient with meta-static breast cancer to the spine will develop clinical symptoms very different from the patient who develops oral cancer on the tongue. The breast cancer patient will almost certainly not present with breast pain; if a breast lump is not noticed by the patient or provider, the initial symptom will likely be pain on movement secondary to metastasis to the skeleton. By contrast, the oral cancer patient would likely have pain during oral function at the very earliest stages of cancer. Furthermore, for the same histologic type of cancer, the symptoms depend on the site of presentation. A patient with squamous cell carcinoma of the lung rarely presents with pain, whereas the patient with squamous cell carcinoma of the oral cavity will almost certainly present with pain as the initial symptom. Thus, animal model studies of cancer pain will ideally be relevant to the histochemical and locational characteristics that have been well-established in the clinical setting. Indeed, Sabino and colleagues have achieved such relevance in an animal model. Specifically, injections of sarcoma, melanoma, or colon adenocarcinoma cells into the distal femur of immunocompromised mice result in varying degrees of bone destruction, central sensitization (as indicated by cFos immunoreactivity and dynorphin expression in the spinal cord), and behavioral indications of pain (3). In this animal model, variation in pain characteristics thus relate to the type of cancer.
Nociceptive Mediators and Modulators
Our review of cancer microenvironmental factors will be confined to those mediators for which there is direct evidence of nociceptive activity. We will not discuss mediators that have been implicated solely on the basis of drug administration [e.g., a COX-2 inhibitor (4)] or techniques directed outside the cancer microenvironment [e.g., downregulation of Toll-like receptor intrathecally (5)]. We also omit from review several nociceptive mediators that are secreted in high levels by certain cancers, including glutamate, cytokines, growth factors, and nitric oxide, because published data do not show that peripheral antagonism of these mediators attenuates cancer-induced nociception. The mediators under review here will be endothelin, protons, proteases, bradykinin, nerve growth factor, and tumor necrosis factor (Figure 1). Endogenous modulators that reverse cancer-induced nociception are also discussed, including cannabinoid receptor agonists.
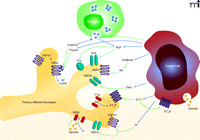
The key cellular components within the cancer microenvironment include the cancer cells, primary afferent nociceptors, and immune cells (e.g., mast cells). Cells comprising the cancer produce and secrete mediators into the cancer microenvironment that modulate nociception. Such mediators include: protons, ET-1, TNFα, NGF, trypsin, and opioids. The cancer can indirectly increase nociceptive mediators such as bradykinin (BK) or tryptase. BK production is increased by the secretion of kallikrein by the cancer cell. Tryptase is released through stimulation of mast cells by the cancer. Certain mediators, such as ET-1 can activate receptors both on the cancer cell (ETBR) and the primary afferent nociceptors (ETAR). The various mediators stimulate the associated receptors on primary afferent nociceptors to produce both nociception and antinociception. (See text for details and references. Blue arrow indicates secretion. Green arrow indicates activation. Red arrow indicates sensitization.)
Endothelin-1: A Dual Role in Cancer-Induced Nociception
The effects of endothelin-1 (ET-1) on cancer pain are unexpectedly complex. The key to understanding these effects is the activity of two endothelin receptor subtypes that differentially affect opioid release from carcinomas. ET-1 is a potent vasoactive peptide that produces nociceptive behavior in animals and humans (6–8) and drives cancer pain (9). Although ET-1 is produced by multiple cancers, it is not produced by all malignancies (10).
ET-1 binds to two G protein–coupled receptors, the endothelin-A receptor (ETAR) and the endothelin-B receptor (ETBR). ETARs are distributed on peripheral sensory neurons; ETBRs are expressed on nonmyelinating Schwann cells of the sciatic nerve and dorsal root ganglion satellite cells (11, 12) as well as on keratinocytes, which are known to secrete opioids (13–15). The ETAR primarily mediates vaso- and bronchoconstriction, mitogenesis, antiapoptosis, and acute pain. ETAR antagonists inhibit osteoblast proliferation and bone metastases proliferation (16–18). The ETBR mediates inflammatory pain and vasodilatation (19, 20).
ET-1 and ETAR Antagonism in Bone Cancer Pain Models
Initial confirmation of the role of ET-1 in cancer pain was presented in two companion reports in 2001 (21, 22). Wacnik and colleagues used a fibrosarcoma bone cancer pain mouse model by implanting fibrosarcoma cells either into the calcaneous bone or subcutaneously adjacent to bone (22). Increased ET-1 levels were characteristic of the whole tumors that developed in these mice, which manifested hyperalgesia. When ET-1 was injected directly into the tumor, moreover, a local nociceptive effect was observed. Although this study did not definitively assign pain behaviors as the direct effect of ET-1 alone within the cancer microenvironment, the authors were able to use ET-1 injection and antagonism to conclude that ET-1 contributes to tumor-induced nociception.
Cain and colleagues used a similar sarcoma model and demonstrated sensitization through behavioral as well as electrophysiologic analysis (21). Hyperalgesia, indicated by ipsilateral paw withdrawal from mechanical stimuli, was again in this study associated with the resulting tumors; specifically, spontaneous activity in C fibers increased with sarcoma progression. The response threshold to heat in the C fibers, but not in Aβ- or Aδ-fibers, of the mice with cancer was significantly lowered. The lack of spontaneous activity in the Aδ-fibers in the sarcoma mouse model contrasts with other work that suggested that subcutaneous injection of ET-1 into the plantar hindpaw excites both C and Aδ-fibers (23).
Intriguingly, the two ET-1 receptor subtypes may mediate opposite effects in models of bone cancer pain. Whereas acute and chronic systemic administration of an ETAR antagonist reduces both ongoing and mechanically evoked pain behavior in a metastatic sarcoma model, an opposite effect is found with ETBR antagonism, which increases pain behavior (12). This duality was similarly reflected in the fibrosarcoma bone cancer pain model discussed above, where injection of an ETAR antagonist directly into the tumor reduced mechanical hyperalgesia, whereas systemic administration of the ETAR antagonist with simultaneous tumor injection of an ETBR antagonist had no effect on the pain behavior (22). This abrogation of the antinociceptive effect of ETAR antagonism by ETBR antagonism has been elaborated and elucidated through the work of Khodorova and coworkers (24) and Quang and Schmidt (25), as described below.
ETAR Antagonism in Soft Tissue Models of Cancer Pain
Using a mouse model that was produced by injecting prostate carcinoma cells into the hindpaw, mechanical hyperalgesia was linked to ET-1 also in soft tissue cancer pain (26). The mechanical hyperalgesia induced by ET-1 could be reversed with oral administration of an ETAR antagonist (26). Another group, also using the hindpaw cancer pain mouse model, subsequently showed that endogenous ET-1 is responsible for cancer-induced nociception (27).
Patients with oral squamous cell carcinoma (SCC), which secretes extremely high levels of ET-1 into the cancer microenvironment (10, 27), report severe functional pain following mechanical stimulation (28, 29). To parallel the mechanical hyperalgesia that is observed in human oral cancer patients, Schmidt and colleagues produced a mouse model of cancer pain by inoculating human oral tongue SCC into the mouse hindpaw. The authors demonstrated that an ETAR antagonist injected directly into the cancer microenvironment produced antinociception similar to that elicited by acutely administered, high-dose, systemic morphine (27). In fact, ET-1 concentration proved to be a more important factor than tumor volume in establishing cancer pain (10). Mice that had been inoculated with melanoma, rather than SCC cells, developed significantly larger cancers but also manifested a higher pain threshold, at all time points, compared to the SCC group of mice
ETAR Antagonists in Clinical Trials
In clinical trials, however, the antinociceptive effect of ETAR antagonists did not hold up. Atrasentan, an orally available ETAR antagonist, has been studied extensively in clinical trials for efficacy in controlling the clinical progression of prostate carcinoma; pain was used as an outcome measure in these studies. Despite the encouraging results of preclinical data, Atrasentan was not shown to significantly reduce cancer pain in clinical trials: among three patient cohorts (placebo, 2.5 mg Atrasentan, 10 mg Atrasentan), no difference was reported in pain (i.e., requirement for opioid treatment) (30). One study demonstrated a trend of pain improvement in a small subset (5 of 15) of patients, but the effect was not significant (31). Most recently, a phase III trial using Atrasentan in 811 men with prostate cancer failed to show that the selective ETAR antagonist significantly reduced pain (32).
ETBR and Opioid Release
The negative clinical trials with the ETAR antagonist indicated that our understanding of the role of ET-1 in cancer pain was far from complete, and the ETBR became central to solving this puzzle. The ETBR had not been as closely studied as the ETAR because the data had been perplexing. For example, upregulation of ETB receptors in cancer cells had been observed in melanoma, breast cancer, and ovarian cancer (33, 34). On the other hand, ETBR downregulation was observed in prostate, bladder, and colorectal cancer (35, 36). ETBR antagonism hindered tumor proliferation in some cases (20, 37), whereas other studies showed conflicting effects on expression. In non-cancerous pain, ETBR mediates both nociceptive and antinociceptive effects of ET-1 (38, 39). ETBR activation was shown to produce an antinociceptive effect in the context of high ET-1 concentration or in the local setting of inflammation (24, 40–42). For example, ET-1 at 10 pmol maximally enhances hyperalgesia following injection of capsaicin into the hindpaw; hyperalgesic enhancement is attenuated at higher doses of ET-1 and is absent at 30 pmol. In addition, preinjection of the ETBR antagonist BQ-788 produces significant hyperalgesia (41), which suggests that the high doses of ET-1 elicit an antinociceptive effect through the ETBR. Further, direct evidence comes from experiments that show that ETBR agonists can completely abrogate ETAR-mediated nociception (41). Electrophysiologic experiments also support the anti-nociceptive effect of ETBR agonism. ET-1 that is applied to cutaneous nerve endings produces action potentials that can be strongly suppressed with ETBR agonists and ETAR antagonists (6, 24).
Most notable is the recent finding that links production of β-endorphin with ETBR activation (43). Specifically, Quang and Schmidt examined mRNA in the SCC cancer pain model and showed that ET-1 expression was nearly doubled, whereas ETBR expression was significantly downregulated in the human oral SCC cell line (compared to normal oral keratinocytes, the non-malignant counterpart to oral SCC). In the mouse model, the intratumor administration of an ETBR agonist attenuated cancer pain by approximately 50% up to 3 hours post-injection, whereas injection of an ETBR antagonist had no effect. Intriguingly, local naloxone methiodide or injection of selective μ-opioid receptor antagonist (CTOP) reversed ETBR agonist-induced antinociception in cancer animals.
A body of circumstantial evidence strongly supports the proposal that peripheral ETBR agonism attenuates carcinoma pain by modulating β-endorphins released from the carcinoma to act on peripheral opioid receptors found in the cancer microenvironment. Oral squamous cell carcinoma consists of malignant keratinocytes that bear ETB receptors and secrete opioids (13–15) to modulate the activity of the surrounding primary afferent nociceptors in skin (44, 45). In addition, ET-1 activation of ETBRs on keratinocytes leads to analgesia that is reversed with naloxone, implicating the keratinocytes as a source of opioid released upon ETBR activation (8, 24). Consequently, the oral cancer mouse model provides intriguing evidence for the potential analgesic role of ETBR activation in carcinomas (25).
ETAR and Opioid Release
Surprisingly, in parallel with the role of ETBR activation, increased production of β-endorphin and increased secretion of leuenkephalin occurs in SCC cell culture treated with ETAR antagonist (25). In the animal model, significant mechanical nociception begins at four days after inoculation of SCC cells and lasts up to eighteen days. Local administration of either naloxone methiodide or selective opioid receptor antagonists (i.e., the μ-opioid receptor antagonist CTOP or the δ-opioid receptor naltrindole, but not the κ-opioid receptor nor-BOR) significantly blocks the antinociceptive effect of the ETAR antagonist.
These results demonstrate that ETBR agonism as well as ETAR antagonism can elicit antinociception through the release of opioid peptides in the cancer microenvironment. Cancers other than oral SCC have also been shown to produce opioids [e.g., malignant melanoma, benign melanocytic naevi (46), small cell lung carcinoma (47), and ovarian tumors (48)] and so modulation by ET-1 receptor ligands could extend beyond the SCC model. Epidermoid carcinoma cells and human foreskin keratinocytes produce proopiomelanocortin (POMC), the precursor for melanotropic, corticotropic, and opioid peptides (49). Opioids secreted by these non-neuronal cells have potentially similar functions as peptides of neural origin. β-endorphins derived from leukocytes can enhance inhibition of inflammatory pain in both humans and animals (50, 51).
The finding that ETAR antagonism leads to opioid secretion and antinociception in the cancer pain mouse model is unexpected, as the generally accepted hypothesis was that antagonism of ETARs present on primary afferent nociceptors elevated the firing threshold (24). In light of the provocative indications of a functional relationship between ETAR and ETBR in the model presented above, data concerning physical associations between ETAR and ETBR are especially intriguing. Specifically, the two receptor subtypes appear capable of forming homo- and heterodimers via coupled binding to the bivalent ET-1 ligand (52). In this way, an ETAR antagonist could disrupt heterodimeric receptor associations, liberating ETBR, which binds to ET-1 with a ninefold increase in affinity (52). Dissociated ETBRs in oral carcinoma cells treated with an ETAR antagonist might thus be more readily activated by ET-1, which is produced in abundance by carcinoma cells, leading to opioid secretion in the cancer microenvironment.
Modulation of ET-1 receptors in the management of cancer pain might have additional benefits, including the control of morphine tolerance. Most types of cancer pain are at least initially responsive to opiates such as morphine, but as tolerance rapidly develops, escalating doses are required, leading to a higher incidence of side effects. ETAR antagonism has been shown to prevent morphine tolerance (53–56). Theoretically, the combination of ETAR antagonism, which produces antinociception and simultaneously prevents morphine tolerance, and ETBR agonism, which leads to local opioid release, might hold promise for the treatment of cancer pain.
Protons and Acid-Sensing Receptors (TRVP1 and ASIC)
A low pH is the hallmark of the cancer microenvironment, reflecting elevated metabolic rates and anaerobic conditions that occur with carcinogenesis. An acidic pH not only activates certain channels, but also sensitizes primary afferent nociceptors, thereby contributing to metastatic cancer pain, one of the most common complications in patients with bone metastases. In this setting, an acidic environment is produced by osteoclasts, which are activated by growth factors secreted by cancer cells, culminating in osteolysis. Acidosis is a well-established cause of pain, and the generation of an acidic microenvironment adjacent to the richly innervated periosteum is a likely mechanism associated with pain in meta-static bone cancer.
A likely nociceptive mechanism for protons in cancer pain is the direct activation of the transient receptor potential vanilloid-1 (TRPV1) channel. TRPV1 is a Ca2+-permeable ionotropic receptor activated by multiple sensory stimuli including heat, acid, and protons. Antagonism of the TRPV1 channel attenuates nociception in a mouse bone cancer pain model, and TRPV1 is present on sensory neuron fibers that innervate the afflicted bone. Acute or chronic administration of a TRPV1 antagonist or genetic disruption of the TRPV1 gene results in a significant attenuation of both ongoing and movement-evoked nociceptive behaviors (57).
The antinociceptive effect of TRPV1 antagonism in a soft tissue cancer model has been demonstrated by injection of SCC into the rat hindpaw (58). The carcinoma that developed within the hindpaw induced significant mechanical allodynia, thermal hyperalgesia, and spontaneous nociceptive behavior, which could be partially ameliorated by morphine. Immunohistochemical analysis showed an increase in the number of TRPV1-positive, large-sized neurons within the dorsal root ganglia. Intraplantar administration of the TRPV1 antagonist capsazepine or the TRP channel antagonist ruthenium red completely inhibited mechanical allodynia and thermal hyperalgesia but did not inhibit spontaneous nociceptive behavior (58).
In bone cancer, the acid microenvironment produces altered expression of acid-sensing ion channels (ASICs), but not of TRPV1 receptors. In a cancer model produced by inoculating MRMT-1 rat breast cancer cells into the tibia of female rats, osteolysis with an abundance of osteoclasts could be detected histologically, and radiographs confirmed bone destruction. c-Fos expression was increased in the spinal cord ipsilateral to site of cancer cell injection, and rats displayed hyperalgesia in the afflicted legs. The bisphosphonate zoledronic acid, which inhibits osteoclast activity, significantly reduced the hyperalgesia and decreased the presence of c-Fos-positive neurons. Expression levels of two ASIC subtypes, ASIC1a and ASIC1b, were increased at the mRNA level in ipsilateral dorsal root ganglions, which could be decreased by treatment with zoledronic acid. ASIC3 and TRPV1 mRNA expression levels, on the other hand, were not elevated in this model (59).
Proteases and Protease-Activated Receptors
Proteolytic activity is critical to carcinogenesis and cancer pain, and the cancer microenvironment is replete with both proteases and proteolytic peptide products (60–62). Cancer-associated trypsin has been identified in cancers such as ovarian carcinoma, pancreatic cancer, hepatocellular and cholangiocarcinomas, lung neoplasms, colorectal cancers, fibrosarcoma, erythroleukemia, gastric cancer, and oral cancer (63). Proteases activate cell surface receptors on primary afferent nociceptors within the cancer microenvironment, either directly or via their peptide products.
Protease activated receptors (PARs) belong to a family of G protein–coupled receptors (PAR1 to PAR4) that are activated by proteolytic cleavage. Such cleavage can result from a number of different enzymes, including serine proteases, trypsin, and tryptase. Cleavage exposes a tethered ligand that binds the receptor and initiates signal transduction (64). PARs can also be activated by short synthetic penta- or hexapeptides that have a sequence similar to the tethered ligand. The peptide sequence SLIGRL activates PAR2 on nociceptors and produces nociception (65, 66), which induces the release of substance P and calcitonin gene related protein (CGRP) from C-fibers in peripheral tissues (67). PAR2 activates multiple second messenger pathways, which sensitize TRPV1 and TRPV4 receptors on nociceptive afferents and result in TRPV1-dependent thermal and TRPV4-dependent mechanical hyperalgesia, respectively (68, 69).
Serine Proteases and PAR2-Dependent Allodynia
PAR2 has recently been implicated in cancer pain by using pharmacologic, behavioral, biochemical, and genetic approaches (70). Proteases capable of activating PAR2 on sensory neurons are recovered in the supernatants of human cancer cells, and human head and neck carcinoma cells manifest increased proteolytic activity. Supernatants from human carcinoma cells can also cause marked and prolonged mechanical allodynia in mice. It is important to note that the injection of supernatant alone, without growth of cancer, induces mechanical allodynia in mice. This nociceptive effect is abolished by serine protease inhibition, diminished by mast cell depletion, and absent in PAR2 knockout mice. The induction of mechanical allodynia by the human carcinoma supernatant is also attenuated by mast cell granule depletion; serine proteases, such as trypsin from cancer cells and tryptase from mast cells, both of which can activate PAR2, contribute to cancer pain. Epithelial cells are likely a secondary source of trypsin (71). Blood vessels surrounding cancers such as gastric carcinoma (72) express trypsin, as do fibroblasts in the surrounding stroma of oral carcinoma (63), and elevated serum trypsin levels have been found in gastric carcinoma patients (73). Chronic exposure to serine proteases secreted by human cancer upregulates PAR2 levels in peripheral neurons (70). The continual release of serine proteases from cancer and non-malignant cells in the microenvironment could produce ongoing excitation of primary nociceptive afferents leading to mechanical allodynia in cancer patients.
Bradykinin
Like endothelin-1, bradykinin (BK) is a vasoactive peptide that plays a role in cancer pain. Certain cancers, such as prostate, secrete kallikrein, which increases the concentration of BK in the cancer microenvironment (74). In a mouse model of bone cancer pain, created by inoculating osteolytic sarcoma cells into the distal femur (75), chronic pharmacologic antagonist blockade (between day 6 and 14 following sarcoma inoculation) of the bradykinin B1 receptor reduced ongoing and movement-evoked bone cancer pain behaviors at both early (10 days post sarcoma inoculation) and advanced stages (14 days post sarcoma inoculation) of bone cancer. The authors demonstrated that chronic administration of B1 antagonist did not affect either tumor proliferation or osteolysis, thereby demonstrating that the B1 antagonist likely has a pure anti-nociceptive effect. This latter determination is important because identification of BK antagonists in antinociceptive mechanisms can be complicated by possible effects on cancer proliferation,
Effects of BK-targeting drugs on pain behavior can differ, depending on cancer histology and site as well as the schedule of drug administration (76). In a skin cancer pain model produced by inoculating melanoma cells into the hindpaw of mice, paw licking in advanced stages of progression, an index of spontaneous nociception, was significantly inhibited by local injection of BK receptor sub-receptor antagonists (specific for either the B1 or B2 type) on day 20 after melanoma inoculation. Mechanical allodynia, on the other hand, was not affected by the B1 receptor antagonist, whereas the B2 receptor antagonist produced a dose-dependent inhibition of mechanical allodynia. Neither of the BK receptor antagonists affected thermal hyperalgesia.
B1 receptors were upregulated in the cancer microenvironment of the skin cancer model. Based on RT-PCR, B1 receptor mRNA was abundant in dorsal root ganglia ipsilateral to inoculation on day 20, but it was very weakly detected on the non-inoculated side; B2 receptor mRNA (detected in the dorsal root ganglia on the non-inoculated side) was not altered by inoculation of melanoma cells. The content of BK and related peptides was increased in the melanoma mass as compared with healthy skin.
Bradykinin Directly Regulates Endothelin-1
Bradykinin studies also add a twist to the complex story of endothelin-1 pain mediation: BK directly induces increased expression and secretion of ET-1 (77). Treatment of cultured melanoma cells with BK increases cellular preproET-1 mRNA levels as well as the secretion of ET-1; the secretion and biosynthesis of ET-1 is regulated through the B2 but not the B1, receptor in this specific melanoma cell line. The effects on ET-1 may not be the same in all melanoma cell lines (20).
Nerve Growth Factor
In the microenvironment of many cancers, sensory neurons are exposed to a chronic increase in nerve growth factor (NGF), which is normally secreted to promote the local growth and survival of afferent sensory neurons. Signals from NGF are mediated via a high-affinity receptor tyrosine kinase (TrkA) and a low-affinity p75 receptor on the neuronal membrane (78). NGF and its high-affinity TrkA receptor can also facilitate proliferation and invasion of multiple cancers, including breast, prostate, and pancreatic cancers (79–84). Expression of NGF and regulation of both high- and low-affinity receptors have also been extensively investigated (85–94). Acute peripheral administration of NGF leads to thermal hyperalgesia (95, 96), whereas chronic administration produces mechanical hyperalgesia (95). Similarly, a transgenic mouse engineered to overexpress NGF exhibits mechanical hypersensitivity (97). Increases in NGF associated with inflammatory and painful conditions occur in keratinocytes in psoriasis (98), synovial fluid in inflammatory joint disease (99), and intestinal tissue in inflammatory bowel disease (100).
Indirect Effects of NGF on Cancer Pain and Implications for Anti-NGF Therapy
NGF also modulates the activity of inflammatory cells, including lymphocytes and mast cells (101, 102); the depletion of mast cells can abrogate nociceptive effects of NGF on small-diameter neurons in a skin-nerve preparation (103). Inflammatory activities can be associated with an increase in NGF, an increase in peptide content in sensory neurons, an increase in substance P and CGRP release from the spinal cord, and an increase in hyperalgesia (104–108). NGF secretion by cancer cells into the microenvironment likely leads to a number of changes that contribute to pain. Chronic NGF exposure leads to an increase in the expression of TRPV1 receptors in sensory neurons (109) and increases ASIC expression and bradykinin receptor binding, both of which contribute to cancer pain (110–114).
One possible mechanism of NGF-induced cancer pain is its association with “perineural involvement,” a pathologic term for the invasion and proliferation of a cancer within a nerve, associated with pain and recurrence following surgical resection. NGF is associated with perineural invasion in adenoid cystic carcinoma, a salivary gland malignancy known for its neurotropism, as well as pancreatic and oral cancer (84, 94, 115, 116). NGF expression is increased in the cytoplasm of pancreatic cancer cells, whereas TrkA is strongly detected in the perineurium of pancreatic nerves but not in the cancer cells. Pancreatic cancers with high NGF and TrkA expression levels exhibit more frequent perineural invasion and higher levels of reported pain. Both NGF and TrkA protein levels are also significantly higher in oral cancers manifesting perineural invasion (94). Anti-NGF can mimic morphine in a mouse model, created through injection of prostate cancer cells into the femur (117), to reduce both early and late stage bone cancer nociceptive behavior. Although NGF clearly has a proliferative effect in cancers, anti-NGF treatment can promote antinociception without influencing tumor-induced bone remodeling, osteoblast proliferation, osteoclastogenesis, tumor growth, or markers of sensory or sympathetic innervation in the skin or bone (117). These results have been taken to indicate that anti-NGF therapy in relieving prostate cancer–induced bone nociception occurs from interference of NGF-mediated sensitization through TrkA and p75 receptors, which are expressed on all nerve fibers that innervate bone.
Tumor Necrosis Factor
Cytokines remain provocative nociceptive candidates because they are produced at high levels in cancer; nociceptive effects of cytokines could be either directly neuronal or immunological. Tumor necrosis factor-α (TNFα) stimulates immune cells, which have the potential to produce nociceptive agents that interact with primary afferent nociceptors in the cancer microenvironment (118). A direct, local role for TNFα in cancer-induced mechanical hyperalgesia has been demonstrated in mice inoculated with mouse fibrosarcoma cells (119). Significantly higher levels of TNFα in this model are detected within tumors, and intraplantar injection of TNFα induces mechanical hypersensitivity (in the fibrosarcoma-established mice as well as in naïve mice). Local production of TNFα likely contributes to tumor-induced nociception, as intra-tumor injection of etanercept, a soluble TNFα receptor, was effective in reducing tumor-induced mechanical hyperalgesia, whereas systemic etanercept was ineffective.
Similarly, Constantin and colleagues used an elegant two-pronged approach to show that TNFα directly induces and maintains cancer-related heat hyperalgesia (120). These authors measured levels of TNFα and other cytokines both in cell culture and in cancer samples from mice inoculated with lung carcinoma cells in the hindpaw. TNFα and IL1β within cancer samples, but not from the cell culture supernatants or cell lysates, were detected at pathophysiological concentrations. Daily systemic treatment with etanercept to neutralize endogenous TNFα abolished cancer-induced heat hyperalgesia (Hargreaves test). The reduction of hyperalgesia was not attributable to any effect on tumor size. To delineate which of the two TNFα receptors (TNFR1 or TNFR2) might be involved in the generation of heat hyperalgesia in the cancer pain model, knockout of the respective receptors was engineered in mice (120). TNFR1−/− cancer mice showed significant heat hyperalgesia, whereas TNFR2−/− mice manifested no heat hyperalgesia until the very late stages of tumor development, suggesting that TNFR2 plays the predominant role in the generation of cancer-induced heat hyperalgesia (120).
Endogenous Mechanisms of Cancer Pain Relief
Cannabinoids have been proposed to be effective against cancer pain for decades (121), but their mode of action in cancer nociception has only recently become clear (Figure 2). Cannabinoids are analgesic in patients with neuropathic pain (122–126) and are antinociceptive and antihyperalgesic in a variety of animal models. Cannabinoids also potentiate the analgesic effects of morphine and prevent tolerance (127, 128). Cannabinoids activate two receptor subtypes, cannabinoid receptors 1 and 2 (CBr1 and CBr2) (129, 130), both of which contribute to analgesia. CBr1 is localized in the spinal dorsal horn, periaqueductal grey (131, 132), and dorsal root ganglion (124, 133). In neuropathic pain, cannabinoids act at central and peripheral nerve CBr1 (125, 126), and at keratinocyte CBr2 receptors (126, 134). Cannabinoid receptors are expressed on nerve terminals and keratinocytes after being synthesized in dorsal root ganglion (135); however, only peripheral CBr1 on nociceptors contribute to antinociception in inflammatory and neuropathic pain models (136). CBr2 is found on immune cells (130, 137) and keratinocytes (134, 138). CBr2 on keratinocytes mediates antinociception via opioid release, similar to the mechanisms discussed above for activation of the ETBR on keratinocytes (134, 138). CBr2 stimulates β-endorphin release from keratinocytes, leading to antinociception through μ-opioid receptors.
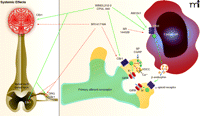
Cannabinoid receptor (CBr) agonists and antagonists act peripherally in the cancer microenvironment on CBr1 and CBr2 subtypes and have central effects when administered systemically. CBr agonists reduce cancer-induced nociception. In the carcinoma microenvironment, CBr1 receptors are found on the primary afferent nociceptive free nerve endings, whereas CBr2 might be present on the carcinoma cells given that they are found on keratinocytes. Activation of peripheral CBr1 reduces hyperalgesia by opening G protein–coupled inwardly rectifying potassium (GIRK) channels, by inhibiting voltage-dependent calcium channels (VDCC), and by inhibiting release of substance P (SP) and calcitonin gene–related peptide (CGRP). An additional mechanism for reducing cancer-induced nociception, including mechanical allodynia, comes from activation of CBr2 on the carcinoma cell, which potentially leads to secretion of beta-endorphins, as occurs in keratinocytes. The endorphins activate mu opioid receptors on the peripheral nociceptive afferent, which open GIRK channels. WIN 55,212-2 and CP 55,940 are non-selective CBr agonists. AM1241 is a selective CBr2 agonist. Selective antagonists include SR141716A for CBr1 and SR144528 for CBr2. The left-hand side of the figure illustrates additional sites of actions for systemic administration of agonists or antagonists of CBr1, namely the dorsal root ganglion cell body, the dorsal horn presynaptic terminals of the primary afferent neuron, and the brain. (See text for details and references. Blue arrows indicate secretion. Green arrows indicate agonism or activation. Red arrows indicate antagonism; red blunt arrows represent inhibition).
Cannabinoid Receptor Agonists Reduce Cancer-Induced Nociception
Cannabinoid receptor agonists can reduce cancer-induced nociception (as measured by grip force) in a mouse model established by inoculating osteolytic cells into the humeri of both forelimbs (139). When peak hyperalgesia was exhibited, WIN55,212-2, a non-selective cannabinoid agonist, was administered intraperitoneally, which elicited time- and dose-related antihyperalgesia. The WIN55,212-2–related change in behavior was confirmed to arise through cannabinoid receptor agonism, rather than as the result of catalepsy and loss of motor coordination, which are well-known side effects of cannabinoids. Because systemic cannabinoids acting through CBr1 are antihyperalgesic in the murine fibrosarcoma pain model (140), various combinations of the nonselective cannabinoid receptor agonist CP 55,940, the CBr1 selective antagonist SR 141716A, and the CBr2 selective antagonist SR 144528 were used to isolate the antihyperalgesic effect of the nonselective cannabinoid receptor agonist to the CB1 receptor.
Cannabinoid-Mediated Antinociception and Opioid Release
To examine the role of peripheral cannabinoid receptors in carcinoma pain, Guerrero and colleagues examined carcinoma-induced allodynia in the oral SCC cancer pain model (10, 27, 141). Intra-tumor administration of either the nonselective WIN55,212-2 or the CBr2-selective agonist AM1241 significantly elevated nociceptive thresholds but functioned within distinct time frames. Both anti-nociceptive activities, however, occurred within twenty-four hours of cannabinoid administration, well before any cannabinoid-associated effect upon tumor growth would have been manifested (141, 142).
In contrast to the carcinoma mouse model, the osteolytic fibrosarcoma hyperalgsesia mouse model exhibits an antinociceptive response to cannabinoid treatment that is solely CBr1-dependent (139, 141). A number of technical differences are likely responsible for the disparate observations. First, fibrosarcoma and squamous cell carcinoma are histologically distinct, and the profiles of nociceptive mediators that they produce are likely different. Second, antinociceptive studies in the two models differed in terms of administration route (local vs systemic) and utilized different ligands to probe for cannabinoid receptor responses.
Regardless of the differences, receptor-selective cannabinoid agonists show significant potential for the management of cancer pain without side effects, and antinociceptive effects in both receptor subtype systems may be opioid-mediated (138, 141). Although systemic, non-selective cannabinoids produce sedation and catalepsy due to CBr1 activation (139), a peripheral CBr2 agonist might provide relief for cancer patients without those side effects.
The diversity of animal models of cancer pain have provided new and important information on the neurobiology underlying cancer pain. Functional interactions between cancer cells and peripheral nerve occur, in part, from the release of several algesic compounds from tumor cells, such as prostaglandins, bradykinin, substance P, cytokines, and growth factors that could excite primary afferent nociceptors in surrounding tissues to produce pain (10, 11, 143, 144). In addition, recent evidence suggests that mechanisms of cancer pain include complex contributions from morphological, neurochemical, and physiological changes in both the peripheral and central nervous systems.
Neurochemistry, Morphology, and Physiology of Primary Sensory Neurons in Cancer Pain
In one of the first animal models of cancer pain, fibrosarcoma cells (NCTC 2472) derived from a spontaneous connective tissue tumor were implanted into the medullary cavity of the femur in C3H/He mice (145–147). Microdialysis revealed high levels of substance P, NGF, and interleukin-10 (IL-10) in the area of the tumor (148). DRG neurons become activated and sensitized in this cancer model, as evidenced by the internalization of substance P receptor in ipsilateral spinal neurons following stimulation (non-noxious as well as noxious) of the femur (145). Importantly, the degree of receptor internalization correlated with the extent of bone destruction. Internalization of substance P receptors in the spinal cord following peripheral stimulation has also been shown in animal models of inflammatory pain (149, 150). Expression of c-Fos, also a marker of inflammation and nociceptor activity and sensitization (151–154), recapitulated the substance P receptor internalization data and similarly correlated with the development of cancer and pain behavior (145). Electrophysiological studies providing direct evidence that nociceptors become sensitized during tumor growth (22) have been discussed above.
Beyond the mechanisms that contribute to nociceptor sensitization in models of bone cancer pain discussed above (e.g., endothelin-1), additional data are developing. Granulocyte- and granulocyte-macrophage colony stimulating factors and their receptors have been implicated (155). It has also been shown that a decrease in endocannabinoid signaling in primary sensory neurons may also contribute to changes in nociceptor excitability produced by fibrosarcoma cells (156).
Tumor growth following implantation of fibrosarcoma cells into the hindpaw also produces morphological changes in peripheral nerve fibers. Morphology of nerve fibers in the epidermis has been investigated because many of these fibers are nociceptors (157, 158). Biopsies obtained from plantar skin of mice that exhibited mechanical allodynia and epidermal nerve indicate sprouting of epidermal nerve fibers early during tumor growth followed by degeneration during tumor progression. Thus, pain associated with this model is due, in part, to nerve injury.
In another model of neuropathic cancer pain, Shimoyama and colleagues implanted Meth A sarcoma cells around the sciatic nerve in mice (159). Mechanical allodynia, thermal hyperalgesia and signs of spontaneous pain developed over several days following implantation. A tumor mass was present around the sciatic nerve but it did not infiltrate into the nerve. Histological evaluation revealed progressive damage to both myelinated and unmyelinated nerve fibers.
Taken together, the studies described above show that tumor growth alters the morphology of peripheral nerve fibers resulting in neuropathy and increased excitability of certain classes of nociceptors. It is likely that the sensitization of nociceptors during tumor development results from a combination of nerve injury, release of inflammatory mediators, and release of algesic substances.
Central Sensitization and Cancer Pain
Mice with bone destruction and hyperalgesia produced by fibrosarcoma cells implanted into the femur exhibit an increase in dynorphin, a pro-hyperalgesic peptide (160), in neurons located in the deep dorsal horn in L3-L5 ipsilateral to the implanted femur, and the expression of dynorphin was correlated to the degree of bone destruction (145). An increase in dynorphin in the spinal cord also occurs in models of neuropathic pain and persistent inflammation (161–163).
Particularly interesting was the increase in labeling with glial fibrillary acidic protein (GFAP), a marker for astrocytes, in the ipsilateral spinal cords of mice with fibrosarcoma-induced cancer pain (145). This increase in GFAP labeling in the dorsal horn was likely due to hypertrophy of astrocytes and also correlated with the extent of bone destruction. Activation of glia in the spinal cord, including astrocytes, may contribute to the development or maintenance of persistent pain by releasing algesic substances that excite or sensitize nociceptive dorsal horn neurons (164).
Although neurochemical changes in dorsal route ganglia and spinal cord occur with many models of persistent pain, including neuropathic, inflammatory, and cancer pain models, the neurochemical changes, rather than being uniform, vary with the particular pain model (154). For example, dynorphin was increased in dorsal horn neurons in mice with hindpaw inflammation and bone cancer, but not nerve injury. Also, mice with bone cancer exhibited a much greater increase in GFAP labeling in the spinal cord as compared to mice with nerve injury, whereas inflammation of the hindpaw did not appear to increase GFAP.
In electrophysiological studies of central sensitization, a model of bone cancer pain in which rat mammary gland carcinoma cells (MRMT-1) are implanted into the tibia of female rats revealed that superficial (lamina I) dorsal horn neurons had enlarged receptive field areas, exhibiting enhanced responses to innocuous and noxious mechanical and heat stimuli. There was also a greater percentage of cells classified as wide dynamic range (WDR), as opposed to nociceptive specific (NS), suggesting that NS neurons were sensitized and behaved functionally like WDR cells. Nociceptive neurons (WDR) in the deep dorsal horn also exhibited greater responses to electrical and heat stimuli applied to the receptive field as compared to controls.
Central sensitization has also been shown following implantation of fibrosarcoma cells into the mouse hindpaw. WDR neurons exhibited an increase in spontaneous activity, and enhanced responses to mechanical, heat, and cold stimuli applied to their receptive field (165). Although the mechanisms that mediate central sensitization following tumor development are not clear, it has recently been shown that mitogen-activated protein kinases may be involved (166).
Conclusion
Cancer and immune cells in the region of tumor masses release several neuroimmune mediators that interact with a variety of receptors on peripheral nociceptive nerve terminals to promote abnormal discharge and hyperexcitability. In addition, tumors growing in the vicinity of peripheral nerves can compromise the integrity of the nerve, inducing a neuropathic condition accompanied by persistent pain, hyperalgesia, or allodynia. Both of these actions of tumors on peripheral nerve can result in central sensitization, which can further enhance the efficacy of nociceptive transmission through the spinal cord dorsal horn and perception of spontaneous and breakthrough pain. Additional studies are needed to further understand the unique molecular mechanisms by which cancer produces sensitization and pain so that new pharmacological targets can be identified that will reduce or block tumor-evoked sensitization.
Acknowledgments
BLS acknowledges support from the National Institute of Dental and Craniofacial Research [Grant DE018561]. DAS and GLW acknowledge support from the National Cancer Institute [Grant CA91007-06].
- Copyright © 2010
References

George L. Wilcox, PhD, is Professor of Neuroscience and Pharmacology, RW Goltz Professor of Dermatology, University of Minnesota Medical School, and Director of the Minnesota Center for Pain Research, University of Minnesota Academic Health Center. His research interests include mechanisms of chronic pain including cancer- and nerve injury-evoked hyperalgesia, novel therapeutic targets for relief of chronic pain, and mechanisms of synergistic analgesic interactions.

Donald A. Simone, PhD, is a Professor and Interim Chair, Department of Diagnostic and Biological Sciences, School of Dentistry, University of Minnesota. Dr. Simone’s research interests include the neural mechanisms that contribute to hyperalgesia, including hyperalgesia produced by tumor growth and chemotherapy, and modulation of hyperalgesia by cannabinoids.

Darryl T. Hamamoto, DDS, PhD, is the Assistant Dean for Academic Affairs and an Associate Professor in the Division of Oral Medicine, Oral Diagnosis, and Oral and Maxillofacial Radiology, Department of Diagnostic and Biological Sciences, School of Dentistry, University of Minnesota. Dr. Hamamoto’s research interests include cancer-evoked hyperalgesia and antihyperalgesic effects of cannabinoid derivatives.

Brian L. Schmidt, DDS, MD, PhD, is a professor in the Department of Oral and Maxillofacial Surgery at the University of California San Francisco. His clinical practice and laboratory research program are closely integrated and are focused on the comprehensive surgical management of head and neck cancer and the peripheral neural mechanisms generating cancer pain. Address correspondence to BLS. E-mail Brian.Schmidt{at}ucsf.edu; fax 415-476-6305.

