A “Wringing” Endorsement for Myosin Phosphorylation in the Heart
Posttranslational modification of proteins influences a myriad of cellular processes including enzymatic pathways, membrane trafficking, and motility–contractility. In skeletal muscle, the phosphorylation of myosin modulates the calcium- (Ca2+ )-dependent regulation of contraction. Evidence provided by Davis et al. (1) of a marked gradient of both myosin light chain kinase (MLCK) expression and activity across the myocardium requires a reexamination of the physiological significance of cardiac protein phosphorylation in vivo.
The myosin II motor molecule of vertebrate striated muscle is a hexamer consisting of two heavy chains (HC, ∼220-kDa) and two pairs of light chains (LC, ∼20-kDa) (Figure 1⇓). The HCs dimerize to form a double-headed, highly asymmetric molecule with head, neck, and tail domains. At the N terminus the HCs form two catalytically active globular heads, each of which contains an ATP- and actin-binding site. Linking the head and tail domains of each HC is the neck, which binds the essential (E-LC) and regulatory (R-LC) light chain subunits. Both LCs are important structural elements of the myosin motor; however, only the R-LC can be phosphorylated (Figure 1B⇓)(2) . Phosphorylation of the R-LC may induce structural alterations in the HC that alter the spatial relationship between the myosin catalytic domain and the thin filament. Electron microscopy of negatively-stained tarantula muscle suggests that phosphorylation of R-LC disorders the helical decoration of myosin heads on the thick filament (3) . Phosphorylation of R-LC may thus “spring” the myosin heads from the thick filament, effectively reducing the inter-filament distance that the myosin cross-bridge must span in order to bind to actin and develop tension.
During the excitation–contraction coupling process, Ca2+ released from the sarcoplasmic reticulum binds to the sarcomeric protein troponin C (TnC). This signal initiates a series of intermolecular movements within the contractile filaments that allows myosin to form a tension-generating complex with actin. Sarcomere shortening results from the ATP-hydrolysis-dependent cycle of cross-bridge attachment and detachment that converts chemical energy into mechanical work. In vertebrate striated muscle, phosphate incorporation by the R-LC modulates this Ca2+ regulation of muscle contraction. For example, stimulation-induced increases in R-LC phosphorylation of mouse and rat fast-twitch muscle correlates with increases in the rate and extent of isometric twitch tension development—that is, a potentiated twitch contraction (4–7) . Phosphorylation of R-LC is also associated with increased work output when mouse fast-muscle shortens against a load during a potentiated twitch (8) . Thus, based on these studies, R-LC phosphorylation appears to augment the force, work, and power of submaximal contractions of mammalian striated muscle.
Data from the study of membrane-permeabilized muscle fibers have provided insight into the molecular basis for R-LC phosphorylation-induced modulations of muscle performance. In these fibers, R-LC phosphorylation increases the Ca2+ sensitivity of the myofibrillar apparatus, thus decreasing the Ca2+ concentration required to elicit a given, submaximal tension (9) . This increased Ca2+ sensitivity arises from a phosphorylation-induced increase in the rate at which cycling cross-bridges make the transition from non-force to force-generating states (10,11) . The mechanism for this effect may be that the addition of a negatively charged phosphate ion to the R-LC displaces the myosin head away from the negatively charged thick filament, thereby increasing myosin head mobility relative to actin and facilitating tension generation (12) . Thus, R-LC phosphorylation-induced increases in the Ca2+ sensitivity of tension development provides a molecular basis for the potentiation of twitch mechanics observed in intact skeletal muscle.
Although a functional role for R-LC phosphorylation in cardiac muscle contraction has yet to be fully established, new work by Davis et al. provides new insights (13) . These authors used submaximally activated skinned fibers to show that, in addition to an increased Ca2+ sensitivity, R-LC phosphorylation alters the stretch activation response—that is, the biphasic tension rise that occurs when a contracting muscle is lengthened. Phosphorylation of cardiac-like slow-twitch fibers decreased the difference between the tension minima that occur when the initial, rapid tension transient relaxes and the tension maxima that occur during the peak of the delayed, slower transient that follows. This dampened contractile response might result from the altered rate at which cycling cross-bridges are able to re -generate tension after perturbation by stretch (10,11). Thus, R-LC phosphorylation mediated modulations of the stretch-activation response may have important implications for the heart, where oscillatory work is performed on a beat-by-beat basis.
In skeletal muscle, MLCK activity is regulated in a Ca2+ -calmodulin-dependent manner (6) . Although cardiac R-LC can be phosphorylated (14–16), evidence for a cardiac isoform of MLCK is generally lacking. Instead, the heart seems to express both smooth and skeletal forms of MLCK (17) . Important in this regard are the results by Davis et al. (1) that shed new light on MLCK expression and R-LC phosphorylation in the heart. These authors show that a skeletal-like MLCK is expressed in a spatially-specific manner across the myocardium, decreasing from the apex to the mid-ventricle region as well as across the heart wall, from the epicardium (large amounts of MLCK) to the endocardium (low amounts of MLCK). Evidence that this MLCK is active in the heart is provided by the pattern of R-LC phosphorylation, which corresponds to the pattern of MLCK expression—again, decreasing from apex to mid-ventricle and from epicardium to endocardium, respectively. This spatial pattern for R-LC phosphorylation may help explain, in part, why epicardial fiber shortening predominates over endocardial fiber shortening during ventricular systole, a force mismatch that produces the “wringing” motion that aids pump function. An R-LC phosphorylation-mediated increase in Ca2+ sensitivity and decrease in the stretch-activation response of epicardial fibers, and the corresponding functional reversal of these parameters in endocardial fibers, may produce the force inequalities that lead to this functionally important torsional response. Given this possibility, future investigations regarding the functional significance for spatially specific distributions of MLCK and corresponding R-LC phosphorylation seem warranted. The prospect of targeting region-specific myosin phosphorylation in the heart may some day represent a new therapeutic avenue for tailoring cardiac hemodynamics in vivo.
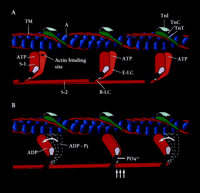
Schematic representation of the contractile and regulatory proteins of the thick and thin filaments of the cardiac sarcomare. A. The actin monomers (A) of the thin filament (blue spheres) and the cable-like tropomysin (TM) molecule (red), as well as the regulatory protein complex consisting of troponin I(TNA)(light blue), troponin C(TNC)(dark green) are shown. The S-I or motor head domain of myosin (red and orange), including the essential light chain (E-LC) (light purple) and the regulatory light chain (R-LC) (purple), is also depicted. S-2, part of the tail domain, oors S-1 to the thick filament.B. The movement of S-1 towards the thin filament proposed to occur following phosphate incorporation(PO4−) by the R-LC is shown(vertical arrows). The force-generating power stroke is energized by the hydrolysis of ATP to its products ADP = P,iand the subsequent release of Pi(curved arrows). Phosphate incorporation by the R-LC may facilitate this force generating event during submaximal Ca2+activation.
- © American Society for Pharmacology and Experimental Theraputics 2002
References

Joseph M. Metzger, PhD, is Professor of Physiology, Professor of Internal Medicine, and Director of the Center for Integrative Genomics at the University of Michigan. His research focuses on genetic modification of normal and diseased heart performance by gene transfer and transgenesis. Address corresponcence to JMM. Email metzgerj{at}umich.edu

Rene Vandenboom, PhD , is a Research Investigator in the Department of Physiology at The University of Michigan. His main interest is the mechanism of muscle contraction. For his doctoral work he studied the modulation of skeletal muscle contraction by myosin phosphorylation. During his postdoctoral years he studied shortening deactivation of skeletal muscle fibers. Currently he is using adenovirus mediated gene transfer to study molecular aspects of cardiac muscle contraction.

