Secretory Functions of Smooth Muscle: Cytokines and Growth Factors
- Address correspondence to WTG. E-mail wtg{at}med.unr.edu; fax 775-784-1620.
Abstract
Smooth muscle cells are important sources of interleukins, chemokines and growth factors in several organ systems. Proinflammatory, mitogenic, and promigratory stimuli produced by vascular and visceral smooth muscle cells may contribute significantly to pathologenesis of atherosclerosis, asthma, inflammatory bowel diseases, and preterm labor. Understanding the role of smooth muscle cells as sources of soluble signals may provide new insights into the cause of these chronic debilitating diseases, and may suggest new targets for developing anti-inflammatory therapy.
Introduction
The primary function of smooth muscle cells is to change, by contracting and relaxing, the shape and stiffness of hollow organs. By relaxing or contracting, smooth muscle regulates blood flow through the vasculature, airflow in the lungs, movement of food and waste through the gut, and uterine delivery of neonates. The neurotransmitters, contractile proteins, and intracellular signaling mechanisms controlling contraction have been studied intensely for many years. We have a good sense of the basic components of the contractile machinery and the regulatory pathways controlling contraction in most autonomic effector organs (Figure 1⇓). It is also clear that smooth muscle cells synthesize extracellular matrix proteins and proteoglycans to fill the extracellular space between muscle cells.
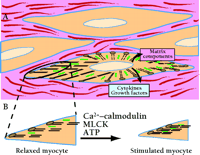
Smooth muscle cells in vivo are embedded in an extracellular matrix consisting of proteins and glycosaminoglycans secreted by smooth muscle cells and fibroblasts. A. Contractile proteins actin (thin filaments; black) and myosin (thick filaments; green) are abundantly expressed and interact to produce force. Under both physiological and pathological conditions, smooth muscle cells also secrete many proteins that probably act as autocrine and paracrine signals. In inflammatory states such as atherosclerosis and asthma, smooth muscle cells are capable of secreting a wide range of cytokines and growth factors into the extracellular space that modify contraction, promote muscle cell growth, and elicit further secretion of signaling proteins. B. Contractility of smooth muscle depends on cellular Ca2+ influx, which activates the myosin light chain kinase (MLCK) reaction that promotes the myosin–actin interactions that underlie contraction. A specific phosphatase (not shown) promotes relaxation of contracted myocytes. See text for details.
In addition to their familiar functions, smooth muscle cells have significant capacity to synthesize, secrete, and respond to small soluble signaling molecules. Data supporting this notion vary among smooth muscle–containing organs; the vasculature has been studied most extensively, although airway smooth muscle and uterine smooth muscle have also received considerable attention. There is considerably less information regarding gastrointestinal smooth muscle. In any case, smooth muscle cells from many organs respond to proinflammatory stimuli, such as interleukin-(IL)-1β , IL-5, IL-13 and tissue necrosis factor-(TNF)-α , by synthesizing a remarkably wide spectrum of signaling proteins including cytokines, chemokines, and peptide growth factors. Smooth muscle cells are thus active, important components of the immune response in the vasculature, the airways, the gastrointestinal system, and the uterus. Figure 2⇓ summarizes the effects of proinflammatory stimuli on the expression of many of these signaling proteins in smooth muscle cells and indicates relevant signal transduction pathways, which involve several mitogen-activated protein kinases; among these are the extracellular signal–regulated kinase (ERK1/2), p38, c-Jun N-terminal kinase (JNK), and the Janus protein tyrosine kinases (JAK). Activation of these kinase pathways by proinflammatory stimuli, as well as by increases in intracellular Ca2+ within smooth muscle cells, activates multiple transcription factors, including nuclear factor-(NF)-κ B, signal transducers and activators of transcription (STATs), nuclear factor for IL-6 expression (NF-IL-6), and nuclear factors of activated T-cells (NFATs).
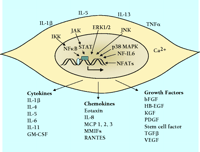
Summary of extracellular stimuli, key signal transduction pathways, and signaling proteins secreted from smooth muscle cells. The extracellular stimuli that appear are examples taken from cell culture studies of vascular, airway, and uterine smooth muscle. IL-1β and TNF-α are the most frequently investigated agents, because they are present at sites of inflammation in the vasculature, the airways, and intestinal tracts. The signal transduction pathways shown are those for which there is evidence in smooth muscle. Many pathways are common to immune cells and are implicated in control of transcription of cytokine and growth factor genes and synthesis of proteins. Each cytokine, chemokine, or growth factor listed has been shown to be synthesized by one or more types of smooth muscle. Some are common to most or all types of smooth muscle, such as IL-1β and bFGF. Others are limited to one particular cell type, such as eotaxin from airway smooth muscle. The functional effects of these agents are varied and in some cases completely different depending on the type of smooth muscle cell.
It is not always clear what purpose is served by the expression of cytokines, chemokines, and growth factors, but there may be several important outcomes. Mediators released from smooth muscle may serve as autocrine and paracrine factors that recruit and retain leukocytes at sites of inflammation. Conversely, secretion of anti-inflammatory cytokines such as IL-10 may serve to limit the magnitude and duration of inflammation. Upregulation of the type 2 angiotensin II receptor (AT2 ) in inflamed vascular smooth muscle (VSM) may serve to limit proliferation and vessel wall remodeling. The net effect on tissue architecture and function will likely be a function of timing and concentration of pro-inflammatory and anti-inflammatory signals. Signaling proteins may trigger remodeling of the smooth muscle and epithelial or endothelial layers of inflamed organs, or they may alter contraction of smooth muscle and thus change organ function. These ideas have important implications for understanding the pathogenesis and treatment of cardiovascular, pulmonary, and gastrointestinal inflammatory diseases.
VASCULAR SMOOTH MUSCLE
The earliest studies of cytokine synthesis in VSM focused on IL-1α , IL-1β and TNF-α (1, 2) . Cultured human VSM cells were treated with bacterial endotoxin, IL-1β , or TNF-α , subsequent to which IL-1 isoforms (mRNA and protein) were assayed. Relatively little basal expression of IL-1 was observed, but significant, rapid synthesis and release occurred in response to proinflammatory mediators. This work stimulated numerous studies in the subsequent decade that verified the initial observations and extended the list of signaling proteins synthesized by VSM. There is now evidence that VSM cells can be induced by a variety of stimuli to synthesize IL-1, -6, -8 and -11 (3–5) , interferon-(IFN)-γ (6) , monocyte chemotactic peptides (MCPs) (4, 7) , macrophage migration inhibitory factors-(MMIFs)-1α and -1β (8) , RANTES (regulated on activation, normal T expressed and secreted) (9) and many peptide growth factors. Platelet-derived growth factor (PDGF) was among the first growth factors shown to be released from VSM (3) . Subsequently, granulocyte/macrophage colony-stimulating factor ( GM-CSF), heparin-binding epidermal growth factor-like growth factor (HB-EGF), basic fibroblast growth factor (bFGF), vascular endothelial growth factor (VEGF), keratinocyte growth factor ( KGF), and hepatocyte growth factor (HGF; scatter factor) were all shown under various conditions to be produced in culture (10–16) .
VSM cells can be “activated” to proliferate, migrate, and secrete matrix proteins in response to many of the signaling proteins designated above. Serum, lipopolysaccharide, IL-1β and TNF-α are the most commonly used stimuli in cell culture systems. Each agent stimulates the production of several signaling proteins, sometimes with important synergism occurring between stimuli. In early studies, IL-1β alone had little effect on VSM proliferation, but when combined with PDGF, growth was enhanced significantly (17) . A similar synergism occurs between IL-1β and bFGF (18) . This pattern of synergism between cytokines and growth factors is reminiscent of that seen in T-cells. Synergism is probably common and highly significant in the microenvironment of the vessel wall, where minute quantities of signaling proteins interact to trigger remodeling of the vessel wall and to regulate VSM contraction.
The effects of secreted signaling proteins on VSM have been investigated extensively. Cytokines and growth factors originating from VSM cells trigger cellular migration, which is likely an early event in vessel remodeling. There is an extensive literature demonstrating promigratory effects for many cytokines and growth factors secreted by smooth muscle cells (see 19, 20 ). Signaling proteins can also alter receptor expression, matrix protein synthesis, contractile protein synthesis, and can further stimulate signaling protein synthesis. The secretion of initial signals, followed by upregulation of signaling protein expression in smooth muscle cells, may thus account for leukocyte migration to VSM. Autocrine and paracrine proinflammatory signaling may thus support the intensive cycles of monocyte and lymphocyte infiltration that can culminate in pathogenesis.
Pathological changes in blood vessel wall structure may also be due to changes in smooth muscle cell receptors and proteins of the extracellular matrix. Inflammation, or proinflammatory treatments of VSM cells in culture, alters expression of receptors for IL-1, endothelin, angiotensin II (Ang II; specifically, the AT2 receptor), vasopressin (specifically, the V1a receptor), and purines (specifically, the P2Y2 receptor) (21–26) . The AT2 receptor is an especially interesting example of a receptor, minimally expressed in the adult vasculature, that is induced by inflammatory events in the vessel wall (24) . The VSM AT2 receptor appears to mediate reduced proliferation and may thereby protect against atheroma formation. On the other hand, Ang II signaling through type 1 receptors (AT1 ) promotes proliferation and atheroma formation; therefore, receptor upregulation in response to inflammation can be either damaging or protective, depending on the signaling system affected.
There are also several examples of signaling proteins that alter the expression and deposition of matrix proteins and proteoglycans. The synthesis of collagen I and III can be increased by transforming growth factor-(TGF)-β and IL-1β (6) , but decreased by IFN-γ . TGF-β and PDGF stimulate the synthesis of proteoglycans, which, by increasing LDL binding in the vessel wall, may be proatherogenic (27, 28) . IL-2 potentiates Ang II–stimulated glycosaminoglycan synthesis (29) . The extracellular matrix thus provides VSM cells a means of modifying the vessel wall in both positive and negative ways.
Several cytokines can also influence VSM contraction. For example, IL-1β and TNF-α inhibit contraction of isolated VSM stimulated with α -adrenoceptor agonists or potassium depolarization (30–33) . IL-1β antagonizes contraction in part by enhancing NO production from smooth muscle cells (34, 35) . Figure 3⇓ summarizes an experiment by Takizawa et al. (35) , in which the phenylephrine-induced (i.e., α -adrenergic) contraction of denuded rat aorta is partially inhibited by IL-1β (Figure 3A⇓). Moreover, the activity of NO synthase is essential to the inhibitory effect of IL-1β , as is apparent upon application of N -monomethyl-L-arginine (LNMMA), an inhibitor of NO synthase (Figure 3B⇓). The cytokine-stimulated production of NO in the vasculature may have both deleterious and protective effects. Specifically, NO activates GTP cyclase to produce cGMP, a second messenger that both decreases intracellular [Ca2+ ] and reduces the phosphorylation of myosin; the ultimate effect of NO is thus to antagonize contraction of VSM cells (Figure 1B⇑). In sepsis, cytokine-elicited NO leads to vasodilation, hypotension, and shock (reviewed in 36 ). On the other hand, NO is also thought to inhibit VSM cell proliferation and reduce VSM cell migration, thereby functioning protectively against atherosclerosis. The net effect of cytokine-induced NO probably depends on the combinations of cytokines and autacoids present, the signaling pathways they collectively regulate, and the duration of cytokine stimulation of VSM cells. A similar argument can be made for cytokine-mediated changes in prostaglandin synthesis. Specifically, an increase in the synthesis of constrictor prostanoids has been implicated in the IL-1β –mediated potentiation of Ang II–induced VSM contraction (37) , whereas IL-6 functions to promote prostacyclin synthesis and thereby indirectly induces the relaxation of vascular rings (38) . Thus, the mechanical effects of cytokines are complex: there may be direct effects on signal transduction in VSM cells; indirect effects arising from NO and prostaglandin synthesis by VSM cells; and indirect effects due to other cell types in the vessel wall, such as endothelial cells and infiltrating leucocytes.
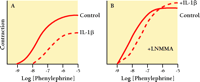
Inhibition of vascular smooth muscle contraction by acute exposure to IL-1β depends in part on nitric oxide synthase activity. Rat aorta strips denuded of endothelium were kept 6 hr in short term organ culture and either left untreated (solid line) or exposed to 20 ng/ml IL-1β (dashed line). Panel A shows IL-1β –inhibited phenylephrine-induced contraction. Panel B shows that when the experiment was repeated in the presence of 100 μ M N -monomethyl-l -arginine (LNMMA) to inhibit nitric oxide synthase, the inhibitory effect of IL-1β was markedly reduced. [Results are summarized from Takizawa et al. (35).]
AIRWAY SMOOTH MUSCLE
An understanding of the signaling proteins that are secreted by airway smooth muscle (ASM) will be crucial in identifying the causes of asthma and developing effective drugs to prevent the narrowing of inflamed airways. In asthma, the contraction of smooth muscle restricts airways that are inflamed, partially occluded by mucous, and narrowed due to the thickening of both the mucosa and the muscularis (39) . Persistent inflammation may chronically activate ASM cells and cause hypertrophy, hyperplasia, and enhanced contraction. The signaling proteins that trigger structural remodeling are thought to come from a variety of cells in the lungs, including T lymphocytes, mast cells, eosinophils, airway epithelium, fibroblasts, and ASM. ASM cells in culture synthesize T helper 1 and 2 cytokines (Th1 and Th2), monocyte chemotactic proteins, and CC chemokines and their receptors (where CC designates the beta family of chemokines, distinguished by adjacent cysteinyl residues) (40–45) . The synthesis of these proteins is commonly elicited experimentally by treatment with either of the acute-phase inflammatory proteins IL-1β and TNF-α , as well as with the combined application of IL-1β plus TNF-α with or without IFN-γ . These agents are used in part because they are commonly found in bronchioalveolar lavage (BAL) fluids from asthmatics, and may thus be assumed to be present in the vicinity of ASM.
ASM cells in culture will synthesize IL-1β in response to IL-1β itself, TNF-α , serum, and combinations of IL-1β , TNF-α and IFNγ (41, 45) . IL-1β also increases IL-6 and IL-8 production (45) , and enhances contraction (reviewed by 46 ). Therefore, IL-1β appears to serve an autocrine function in airways, with multiple functional effects on ASM.
ASM cells in culture synthesize several Th2 cytokines, including IL-5, IL-6, and GM-CSF. IL-5 is also synthesized in isolated ASM in response to serum from atopic asthmatics (47) . GM-CSF is a growth factor, present in BAL fluids of asthmatics, that supports proliferation and survival of eosinophils and neutrophils (43) ; it is thus thought to be a prominent factor in the eosinophilia that develops in the lower airways of asthmatics (48) . ASM cells in culture synthesize GM-CSF in response to IL-1β and TNF-α (49) . IL-6 and IL-11 are produced in ASM in response to IL-1β , TNF-α , and TGF-β (50, 44, 45) . IL-6 manifests several effects on lung cells that would exacerbate asthma, including effects on B-cell, T-cell, and mast cell function and differentiation, as well as increased mucous secretion. In addition, IL-1β and IL-6 are mitogens of cultured ASM (51) , and some of the effects of IL-1β may occur indirectly via production of IL-6. IL-11 was one of the first cytokines shown to be synthesized by ASM, but little is known of its functional effect on ASM cells (50) .
Chemokines synthesized by ASM include the CC chemokines RANTES, monocyte chemoattractant proteins (MCPs) 1, 2 and 3, and eotaxin, as well as the CXC chemokine IL-8. RANTES and MCPs are produced in response to IL-1β , TNF-α and IFN-γ (52, 53, 54) . Eotaxin is produced in culture in response to IL-1β and TNF-α (54, 55) . These chemokines are found in BAL fluids of asthmatics and serve as chemoattractants for leukocytes. Eotaxin attracts eosinophils into the airways and thus promotes the eosinophilia characteristic of atopic asthma. The notion that smooth muscle cells are a significant source of this chemokine is strongly supported by in situ immunostaining of ASM tissue from asthmatics (55). IL-8 is also a chemoattractant for eosinophils that promotes neutrophil degranulation. IL-8 synthesis is increased by bradykinin (56) and by IL-1β , TNF-α , and IFN-γ (57, 53, 45) .
The effects of signaling proteins on ASM are often similar to the effects on VSM. Growth factors and some cytokines are mitogenic and are thought to contribute to hyperplasia of the airways of asthmatics (58) . IL-1β and IL-6 elicit both hyperplasia and hypertrophy of ASM cells in culture (51) . ASM cells also migrate in vitro in response to several mediators, including IL-1β , PDGF and TGF-β (59) . Proliferation and migration of VSM cells are thought to be a prominent and important responses to proatherogenic and vasculoneogenic signals. ASM cell proliferation must occur at some modest rate during remodeling of asthmatic airways, but whether cell migration is necessary for smooth muscle thickening is an open question.
Cytokines and growth factors alter contraction elicited by acetylcholine, histamine, and tachykinins in a manner consistent with the narrowing of airways in asthma. Signaling proteins that modify contraction include IL-1β , -2, -4, -5, -8, -10 and -13, TNF-α , TGF-β and IFN-γ (see 60 ). IL-1β potentiates contractile agonists and antagonizes β -adrenergic relaxation (61–63) . This is in contrast to marked inhibition by IL-1β of the α -adrenergic contraction of VSM (Figure 3⇑).
There are also examples of interesting paracrine interactions of cytokines that affect ASM contraction. IL-5, an early signal in development of atopic asthma, potentiates contraction of rabbit ASM elicited by acetylcholine (41) . Hakonarson et al. (47) showed that isolated ASM passively sensitized by serum from atopic asthmatics manifests proasthmatic characteristics that depend on the production of IL-5, and that the IL-5 triggers the autologous production of IL-1β . IL-1β increases contraction induced by acetylcholine and tachykinins and reduces sensitivity to β -adrenergic agonists. The mechanism(s) by which IL-1β modifies contraction include altered G protein expression, upregulation of cyclooxygenase-2 expression, and increased prostaglandin E2 production. The predominant effect of IL-1β in studies of isolated ASM in vivo is a marked inhibition of β -adrenergic relaxation (64) , which may underlie the well-known resistance to β -agonists seen in some asthmatics.
TNF-α potentiates ASM contraction elicited by muscarinic receptors and bradykinin, probably by increasing intracellular calcium (65) and enhancing calcium sensitivity of contractile proteins (66) . In contrast to enhanced contraction and exacerbation of airway narrowing by TNF-α and IL-1β , IL-2 and IFN-γ may have a beneficial effect in asthmatic airways by inhibiting smooth muscle contraction. These agents reduce contraction elicited by cholinergic stimulation of passively sensitized ASM (41) .
Further in vivo studies are required to define the net effect of cytokines on ASM contraction. ASM in asthma is exposed to elevated levels of bronchoconstrictors including acetylcholine, histamine, and leukotrienes, which often act synergistically. Contraction may be further increased, and relaxation inhibited, by one or more cytokines present during inflammation. Smooth muscle cells of the airways may be a primary source of the cytokines producing inappropriate bronchoconstriction.
UTERINE SMOOTH MUSCLE
Interest in intercellular signaling mechanisms controlling the onset of labor has stimulated several surveys of signaling proteins released by cultured human myometrial cells. Synthesis of IL-1β , -6, and -8 in response to proinflammatory signals have been described (67) . Immunomodulatory proteins TNF-α and GM-CSF are also synthesized by normal myometrial cells (68, 69) , and several peptide growth factors, including PDGF (70) , bFGF (71) , epidermal growth factor (EGF) (72) , insulin-like growth factor-(IGF)-1 (73) , and TGF-β (74) , have been shown to be released by myometrial smooth muscle. These growth factors stimulate smooth muscle cell proliferation and probably contribute significantly to uterine tissue remodeling during pregnancy.
In addition to promoting proliferation, secreted signaling proteins also trigger changes in uterine smooth muscle receptor, hormone, and prostaglandin production. The response of uterine smooth muscle to secreted IL-1β includes increased expression of oxytocin and oxytocin receptors (67, 75) . Oxytocin expression is also increased by IL-6 (76) . PDGF stimulates production of VEGF (77) , which may contribute to angiogenesis in the pregnant uterus. Similar to both vascular and ASM, uterine smooth muscle cells upregulate cyclooxygenase-2 and prostaglandin E2 receptors in response to IL-1β , consistent with the notion that cytokines activate uterine smooth muscle at term to initiate parturition (78) . Indeed, IL-1β induces contraction of rabbit uterine smooth muscle, and the contraction is potentiated by IL-8 (79) , although IL-8 alone has no contractile activity; potentiation of cytokine-induced smooth muscle contraction by a second cytokine thus emerges as a common theme. Synergism between cytokines, hormones, and prostaglandins has important implications for uterine smooth muscle during parturition, when multiple signaling proteins appear to coordinate optimal uterine contraction. Interestingly, prolonged exposure to IL-1β inhibits the levels of oxytocin signaling seen upon acute exposure to IL-1β , and suggests that preterm labor triggered by infection and persistent IL-1β elevation may not be mediated by oxytocin (80) . Although not as extensive as the cardiovascular literature, data suggest that uterine smooth muscle cells produce numerous signaling proteins with significant, various functional effects on pregnancy, inflammatory processes in the uterus, and initiation of parturition.
GASTROINTESTINAL SMOOTH MUSCLE
There is an extensive literature on the production of cytokines and growth factors by the intestinal mucosa, as well as by invading leukocytes in inflammatory bowel diseases. In contrast, there are relatively few reports that gastrointestinal smooth muscle cells per se are sources of signaling proteins. One recent study of colonic smooth muscle from patients with ulcerative colitis demonstrated the expression of IL-1β in the muscularis (81) , which is consistent with a previous report of IL-1β expression in a rat model of colitis (82) . IGF-1 and IGF receptors are also upregulated in smooth muscle during experimental colitis, which might indicate remodeling of the smooth muscle layer in response to inflammation (83) . A developmental study suggests cells of smooth muscle lineage produce stem cell factor, a ligand for c-Kit, which is a tyrosine kinase important in development of interstitial cells of Cajal (84) . It is interesting that stem cell factor is produced also by airway, vascular, and uterine smooth muscle, and that it is implicated in the recruitment and survival of mast cells in the airways (29, 85, 86) . A similar function for smooth muscle–derived stem cell factor in the gastrointestinal tract seems quite possible. Given the environment that exists in the wall of the colon during inflammation, it would be surprising if smooth muscle cells did not respond by synthesizing many or most of the signaling molecules described in other types of smooth muscle; however, results are limited at this point to IL-1β and a few growth factors.
Studies of functional effects of cytokines and growth factors on gastrointestinal smooth muscle cells are also somewhat limited. Exogenous EGF and bFGF are effective mitogens used to culture colonic smooth muscle cells (87) . IGF-1 and bFGF are localized to the muscalaris of the gut (71) , but it is not known whether smooth muscle cells synthesize the wide range of peptide growth factors seen in other organs. It is also not known what effects cytokines have on gastrointestinal smooth muscle contraction. One of the few published reports shows that IL-1β potentiates contraction of isolated rat stomach smooth muscle (88) , which is consistent with the stimulatory effects of IL-1β on uterine smooth muscle and ASM, but contrasts with inhibition of VSM contraction. The relevance of IL-1β or any other cytokine to enhance the smooth muscle contraction that occurs in response to clinical conditions of paralytic ileus, various types of colitis or ileitis is unclear. In those conditions, the net effect of inflammation on overall gastrointestinal motility is inhibitory, probably through inhibition of neurotransmission (89, 90) . Signaling proteins may enhance contraction in isolated muscle preparations, but the net effect in vivo will be the integrated response to both excitatory and inhibitory stimuli acting on smooth muscle cells, interstitial cells, and enteric neurons.
CONCLUSION
Smooth muscle cells are metabolically dynamic, with the potential to express and secrete numerous, highly active signaling proteins. The secretory function appears to be a common feature of all smooth muscle–containing organs. The course of several common and highly debilitating chronic diseases, such as atherosclerosis, asthma, inflammatory bowel diseases, and interstitial cystitis, as well as the onset of labor, may be determined in part by the sets of proteins secreted from smooth muscle cells. Two new lines of investigation are needed to define the significance of smooth muscle–derived cytokines and growth factors: More in situ assays will be needed to demonstrate where, when, and to what extent signaling proteins are secreted, especially in the airway, gastrointestinal tract, and uterus. It will also be important to define the extent of bidirectional paracrine cytokine and growth factor signaling between smooth muscle cells, immune cells, and nerves. New studies are also needed to define more completely the signal transduction pathways in smooth muscle cells that are necessary and sufficient for the secretion of signaling proteins. The effect of current anti-inflammatory and anti-proliferative drugs on these signaling mechanisms in smooth muscle is a topic of great interest, and development of new drugs targeting the secretory function of smooth muscle is a topic with great potential.
Acknowledgments
The authors gratefully acknowledge the support of the National Heart Lung and Blood Institute and the National Institute of Diabetes and Digestive and Kidney Diseases.
Footnotes
-
William T. Gerthoffer, PhD, is Professor of Pharmacology at the University of Nevada School of Medicine.
-
Cherie A. Singer, PhD, is currently Research Assistant Professor of Pharmacology at the University of Nevada School of Medicine.
- © American Society for Pharmacology and Experimental Theraputics 2002

