Mechanisms of Synaptic Plasticity: From Membrane to Intracellular AMPAR Trafficking
Current interest in synaptic plasticity, or alterations of neuron-to-neuron communication efficacy, stems from the belief that plasticity underlies key aspects of higher levels of cognitive function. The most widely studied models of synaptic plasticity are long-term potentiation (LTP) and long-term depression (LTD), which reflect the sustained modifications of synaptic strength after brief periods of repetitive synaptic activity. It is generally believed that clarification of the cellular and molecular mechanisms for LTP and LTD will aid our understanding of neural development, adaptation, learning, and memory. In current models, brief periods of repetitive synaptic activity lead to sustained changes in synaptic transmission. Critical events in the formation of plasticity include opening of N- methyl-d -aspartate (NMDA)–sensitive glutamate receptors (NMDARs), increases in postsynaptic calcium concentrations during repetitive synaptic activity, and activation of a variety of kinases, including calcium-calmodulin–dependent protein kinase II (CaMKII) (1), mitogen-activated protein kinase (MAPK) (2), the tyrosine kinase Src (3), and phosphatidylinositol 3′-kinase (PI3K) (4).
Synaptic activity can lead to the regulated trafficking of postsynaptic α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)–sensitive glutamate receptors (AMPARs) at excitatory synapses during the plasticity phase (5–8). The AMPARs are multimeric proteins composed of the subunits GluR1, GluR2, GluR2L, GluR3, and GluR4 (9–11). The cytoplasmic C termini of AMPAR subunits, which exist in either long or short forms, determine the trafficking characteristics of AMPARs (Figure 1⇓). AMPARs with long cytoplasmic tails (e.g., GluR1, GluR2L, or GluR4) are normally restricted from synapses, but are delivered to synapses during activity-induced synaptic enhancement (4, 12, 13). The synaptic delivery of GluR1- or GluR2L-containing AMPARs requires the activation of NMDARs and CaMKII. However, NMDAR-dependent delivery of GluR4-containing AMPARs to synapses does not require CaMKII activity. Because GluR4 is expressed earlier in development than GluR2L and GluR1 (12), synaptic delivery of GluR4-containing AMPARs is likely to be responsible for LTP in the neonatal animal, whereas synaptic delivery of GluR2L- and GluR1-containing AMPARs is responsible for LTP in the juvenile and adult (14). The newly delivered AMPARs with long cytoplasmic tails will eventually be replaced by AMPARs with short cytoplasmic tails (e.g., GluR2 or GluR3), a process that does not require synaptic activity (12). Short-tailed AMPARs shuttle continuously from nonsynaptic to synaptic sites and replace synaptic receptors without changing synaptic efficacy (4, 13) ; the total number of short-tailed AMPARs in this cycling pool, including those at synaptic and nonsynaptic sites, can be reduced after activity-induced synaptic depression (13, 15–19).
Recent studies have begun to reveal the biochemical pathways linking NMDAR activity with AMPAR trafficking. There is good evidence indicating that CaMKII acts as the downstream sensor of synaptic activity and facilitates NMDAR-dependent LTP (1, 7). For example, CaMKII, activated during LTP, enhances synaptic transmission by promoting the delivery of AMPARs into synapses, and this enhanced synaptic transmission presumably exhausts all available insertion sites for long-tailed AMPARs at synapses, thus occluding further induction of LTP. Finally, pharmacological or genetic inhibition of CaMKII activity blocks LTP. The molecule that relays NMDAR activity and CaMKII signaling and controls synaptic delivery of AMPARs is the small guanosine triphosphatase (GTPase) Ras. In contrast, Rap, another member of the Ras GTPase superfamily, relays NMDAR activity and controls synaptic removal of AMPARs. Support for these conclusions comes from several lines of studies. First, Ras, Rap, and their activators [guanine nucleotide exchange factors (GEFs)] and inactivators [guanosine triphosphatase–activating proteins (GAPs)] are located at synapses, where they control important neuronal functions and have significant effects on behavior (20–22). In addition, inhibiting the Ras signaling pathway blocks CaMKII-induced synaptic potentiation and LTP, whereas a constitutively active Ras signaling pathway mimics CaMKII-induced synaptic potentiation by driving synaptic delivery of AMPARs with long cytoplasmic tails; this synaptic enhancement occludes CaMKII-induced synaptic potentiation and LTP (19). Finally, inhibiting the Rap signaling pathway blocks synaptic removal of AMPARs and LTD, whereas a constitutively active Rap signaling pathway removes all synaptically located short-tailed AMPARs; this synaptic depression occludes further induction of LTD (19). Because Ras increases phosphorylation of p42/44 MAPK [(the extracellular-regulated kinase (Erk)1 and Erk2], whereas Rap increases phosphorylation of the p38 MAPK (19), it is concluded that the Ras-Erk1/Erk2 signaling pathway links NMDAR activity with synaptic delivery of AMPARs by relaying CaMKII signaling, and the Rap–p38 MAPK signaling pathway links NMDAR activity with synaptic removal of AMPARs (Figure 1⇓). These results are consistent with the findings that genetic mutations in of molecules in the Ras or Rap signaling pathways cause diseases with cognitive impairment (e.g., autism, neurofibromatosis, tuberous sclerosis, and X-linked mental retardation) (22–25).
Many studies have focused on AMPAR trafficking and regulation at synapses; however, little is known about the intracellular trafficking character of AMPARs before they arrive at synapses. An elegant study by Greger et al. now reveals that egress of AMPARs from the endoplasmic reticulum (ER) is also tightly regulated in neuronal cells (26). As is the case with AMPAR trafficking at synapses, the cytoplasmic C-terminal tails of AMPAR subunits appear to define the rules governing intracellular AMPAR trafficking (Figure 1⇓); however, the rules are opposite to those at synapses. AMPARs with long cytoplasmic tails can mature and leave the ER rapidly, but AMPARs with short cytoplasmic tails are largely confined within the ER; they mature and travel from the ER at a much slower rate. The C terminus of GluR2 is required for AMPARs to mature and exit from the ER. More importantly, the major regulation element is the Q/R–editing site in the GluR2 channel pore region: In GluR1, GluR3, and GluR4, there is a glutamine (Q) at this critical pore site, whereas in GluR2, the uncharged glutamine is replaced by a charged arginine (R). Reversion of the mutation at this site (R607Q) in GluR2 mimics GluR1-like egress from the ER. Thus, Greger et al. propose that, like AMPARs with long cytoplasmic tails at synapses (cf. 7), AMPARs with only short cytoplasmic tails are normally kept within the ER by a retention factor (26). The retention of long-tailed AMPARs seems to be suppressed, so that these receptors can leave the ER more freely.
It is likely that trafficking of AMPARs from the ER can modulate synaptic plasticity by controlling the size of AMPAR pools. Previous studies have demonstrated that altering the pool size of AMPARs at nonsynaptic (cytosolic) sites can influence the synaptic delivery of AMPARs (12, 19). Thus, the rate at which AMPARs with long cytoplasmic tails leave the ER may modulate activity-dependent delivery of AMPARs with long cytoplasmic tails and LTP. On the other hand, AMPARs with only short cytoplasmic tails cycle continuously between cytoplasm and cell membrane (synapses), and the strength of synaptic transmission mediated by these receptors is determined by the total number of the receptors available in this cycling pool (4, 13, 15). Indeed, Rap activity induces synaptic depression and LTD by removing AMPARs out of this cycling pool (19). Because short-tailed AMPARs will enter into the cycling pool after exiting from the ER, the newly identified rule that governs their egress from the ER may provide additional regulation for the size of the cycling pool of AMPARs and LTD. Further study is required to determine whether egress of AMPARs from the ER with long cytoplasmic tails modulates LTP and whether egress of AMPARs with only short cytoplasmic tails regulates LTD.
The study by Greger et al. (26) raises many other questions. For example, the biochemical cascades that trigger the egress of AMPARs have yet to be identified. It is known that the phosphorylation of NMDARs is required for the exit of NMDARs from the ER (27). Whether Ras and Rap signaling pathways, which control synaptic AMPAR trafficking, are also involved in signaling AMPAR trafficking is unclear. Given that the methodology for studying intracellular AMPAR trafficking has now been established by Greger et al. (26), we expect that these signaling pathways will soon be identified and characterized. Understanding the signaling pathways regulating AMPAR trafficking would certainly lead to a better understanding of the cellular and molecular mechanisms of synaptic plasticity and suggest additional molecular targets to which novel genetic and pharmacological therapies could be designed to treat insidious mental diseases.
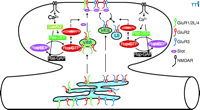
Models for synaptic and intracellular AMPAR trafficking. At synapses, AMPARs (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors) with long cytoplasmic tails (GluR1/2L/4) are retained at nonsynaptic (cytoplasmic) sites and only inserted into synapses after Ras activation, whereas AMPARs with only short cytoplasmic tails (GluR2/3) continuously cycle between synaptic and nonsynaptic sites and they can be removed from the cycling pool after Rap activation. At the ER, AMPARs with long cytoplasmic tails exit from ER rapidly whereas AMPARs with only short cytoplasmic tails are retained in the ER and only exit from ER after Q/R—editing. AMPAR, AMPA receptor; ER, endoplasmic reticulium; LS, lysosome; V/ES, vesicles-endosomes.
- © American Society for Pharmacology and Experimental Theraputics 2003
References

J. Julius Zhu, PhD, is an Assistant Professor in the Department of Pharmacology of the University of Virginia School of Medicine, an Investigator at the Ronald and Nancy Reagan Research Institute of the Alzheimer’s Association, and a Naples Investigator of the National Alliance for Research on Schizophrenia and Depression (NARSAD) foundation. Email jjzhu{at}virginia.edu; fax 434-982-3878.

