Sex, Digitalis, and the Sodium Pump
Digitalis-like steroids and related agents have been a mainstay in the treatment of congestive heart failure (CHF) ever since the publication, in 1785, of Withering’s seminal monograph on the foxglove (1) . CHF refers to the clinical syndrome that results when the heart is unable to pump sufficient blood to keep up with the metabolic demands of the body. CHF is characterized by excessive neuronally and hormonally mediated fluid retention, expanded intravascular volume, high pulmonary and systemic venous pressures with consequent dyspnea (shortness of breath) on exertion, reduced exercise tolerance, and fatigue. Most CHF patients also have impaired ventricular systolic function and depressed cardiac output; these are the patients most often treated with digitalis glycosides.
Our understanding of the mechanism of action of digitalis has continued to evolve since the discovery (2) that they are positive inotropic agents (i.e., they are “cardiotonic” and enhance cardiac contraction). The digitalis glycosides, sometimes referred to as “cardiotonic steroids” (CTS) cause cardiac muscle to lose K+ (3) and gain Na+ because all CTS inhibit the Na+ pump (Na+ , K+ –ATPase) (4) , a plasma membrane (PM) protein, present in all cells, that consists of α and β subunits. The Na+ pump uses the energy from ATP to extrude Na+ and to maintain the large Na+ electrochemical gradient across the PM.
But how does inhibition of the Na+ pump augment cardiac contraction? Discovery of the Na+ -Ca2+ exchanger (NCX) (5, 6) provided the missing link between Na+ pump inhibition and delivery of Ca2+ to enhance contractility (6) . Indeed, prevention of NCX expression abolishes the cardiotonic and vasotonic (increased vascular contractility) effects of CTS such as ouabain (7, 8) .
The dogma of NCX mediated CTS activity, described in most pharmacology textbooks (9) , was complicated by the discovery of several isoforms of the Na+ pump in the 1980s (10) . Molecular cloning identified four isoforms of the catalytic (α ) subunit (11) , which contains the Na+ , K+ , ATP and CTS binding sites. Most cells express α 1 as well as α 2 or α 3; α 4 has been detected only in sperm (11) . The α isoforms are disinguishable by their regulatory and kinetic properties (12, 13) , and by their cellular distribution (14) . For example, α 1 has the highest affinity for Na+ (13) and is uniformly distributed in the PM (14) ; this “housekeeping” isoform maintains the low total cytosolic Na+ concentration ([Na+ ]CYT ). In contrast, isoforms α 2 and α 3 have lower affinities for Na+ (13) and, at least in some cell types, appear to be confined to NCX-containing microdomains in the PM that are specifically juxtaposed to sarcoplasmic or endoplasmic reticulum (“junctional” SR or ER) (14–17) . Thus, the α 2 and α 3 isoforms are contained within PM-junctional SR–ER units (“PLasmERosomes”) that function as Ca2+ -signaling complexes (CaSCs), as diagrammed in Figure 1⇓. Isoforms α 2 and α 3 are the primary targets of low-dose CTS; selective reduction of the activity of these isoforms enhances Ca2+ signaling in heart, vascular smooth muscle, and other cell types (16–21) . This implies that the Na+ pump α 2 or α 3 isoforms, by controlling NCX, critically influence the Ca2+ concentration in junctional SR–ER and, thus, Ca2+ signaling (Figure 1⇓) in many types of cells. These multiple possible sites of CTS action may be relevant to the therapeutic effects of digoxin in heart failure which include reducing heart rate, sympathetic nervous system activity and renin activity, all of which are overactive in CHF (22, 23) .
Another complication that is not usually taken into account in assessing the in vivo cardiotonic activity of digitalis glycosides is the presence of endogenous CTS, including endogenous ouabain (EO) in the human circulation (24, 25) . EO is synthesized and secreted by the adrenal gland (26) ; small amounts also may be produced in the brain (27) . Indeed, elevated circulating levels of EO are found in approximately half the patients with CHF, in whom the EO level is inversely correlated with cardiac index (28) . This suggests that EO secretion is increased as a compensatory response in some CHF patients.
In 1997, the Digitalis Investigative Group (DIG) reported on a controlled, randomized trial of digoxin in 6,800 patients with CHF already being treated with diuretics and angiotensin converting enzyme (ACE) inhibitors (29) . The study led to the conclusion that digoxin therapy reduces hospitalization for worsening heart failure and confirmed the overall efficacy of digoxin therapy in patients with heart failure and normal sinus rhythm. Yet, for unknown reasons, digoxin benefits only some patients. Clearly, some means of identifying the potential impact of digoxin prior to its administration would be extremely useful. One possibility relates to the elevated circulating levels of EO (1–9 nM) in many CHF patients (28) that overlap the therapeutic range for digoxin and ouabain (30) . The sensitivity of human cardiac Na+ pumps to digoxin and ouabain is similar (31) . Thus, the plasma EO concentration might affect the response to digoxin. For example, a high EO level might predispose patients to digitalis toxicity. Conversely, patients with low plasma EO might be more likely to benefit from long-term digoxin therapy. It is unfortunate, given the unprecedented scale of the DIG trial (29) , that endogenous CTS were not considered during its design and implementation.
In a retrospective analysis of data from the DIG study, Rathore et al. (32) examined the effect of gender on the outcome of patients with CHF who were treated with digoxin. They observed that digoxin therapy significantly increased the risk of death from any cause in women, but not men, with heart failure and left ventricular systolic dysfunction. In an accompanying editorial, however, Eichhorn and Gheorghiade (33) noted that, after one month of therapy, the women had “significantly higher” serum digoxin levels than did the men (0.9 versus 0.8 ng/ml; p = 0.007), despite the administration of lower doses of digoxin. Because digoxin has a narrow therapeutic window, this seemingly small difference raised the possibility that the higher serum digoxin levels in women may have increased the risk of arrhythmias. These authors recommended that therapy with digoxin should not be discontinued, but should be employed at a dose that maintains the serum digoxin concentration below 1 ng/ml.
Close inspection of the Figure 1⇓ data of Rathore et al. (32) reveals, however, that the reason for the gender effect is an apparently lower mortality rate in placebo-treated women than in digoxin-treated women or in both placebo- or digoxin-treated men, starting at about twelve months after the start of treatment. Because the plasma digoxin levels in men and women were not different at this twelve month time point, speculation that a high plasma digoxin concentration, per se, was the cause of the excess mortality in treated women (32) seems less justified. Because 88% of the women, but only 80% of the men (p < 0.001) were also on diuretic therapy (32) , another possibility is that more women may have had hypokalemia (lower than normal levels of circulating K+ ); cardiac arrhythmias may then have been exacerbated by digoxin treatment.
There are no specific gender-comparisons of EO concentrations that might shed light on the gender effect of digoxin, nor are there reports that explicitly compare gender differences regarding the sensitivity of human Na+ -pump isoforms to CTS. It is conceivable that the excess mortality could reflect a mechanism that depends not only on plasma digoxin or EO concentration, but also on estrogen-dependent and other gender-related factors or mechanisms that affect cellular Na+ and/or Ca2+ handling. For example, women appear to have lower Na+ concentrations and fewer Na+ pumps in their erythrocytes than do men (34) , and women with CHF may also have slightly fewer skeletal-muscle Na+ pumps (35) . Although it is not clear whether these observations accurately extrapolate to cardiac myocytes or neurons, the implications could be significant in the present context: at comparable circulating levels of digoxin, there would be fewer active PM-localized Na+ pumps in women. In the pre-existing condition of heart failure, additional stress could lead to reduced reserve capacity of myocardial Na+ pumps thereby predisposing women to potentially fatal arrhythmias.
Another important issue concerns hormone-replacement therapy (HRT) because the median age of women in both the digoxin and placebo groups was sixty-six years and most of them were surely post-menopausal. Although it is likely that many of the women were on HRT, these data were not collected in the DIG study (32) . A post hoc analysis of data from the Heart and Estrogen/Progestin Replacement Study (HERS) (36) may, however, be relevant. Analyses of HERS data revealed that among digoxin-treated women, those on HRT had a significantly higher incidence of coronary events (nonfatal myocardial infarction or death from coronary heart disease) during the first year of the four-year study. Among women who did not receive digoxin, there was no HRT-related difference in the incidence of coronary events. Because the digoxin treatment in these subjects was not randomized, an unresolved question is whether the women who received digoxin therapy were sicker than those who did not.
In the context of HRT, it is noteworthy that the administration of CTS increases circulating estrogen levels (37, 38) ; moreover, CTS may, themselves, have weak estrogenic activity (39) . Therapy with digitalis has been reported to induce gynecomastia (breast enlargement) and increase the risk of breast cancer in men (40) , but reduce the mortality rate in women with breast cancer (41) .
The effects of estrogen (and testosterone) on Na+ pumps in experimental animals are controversial: there are reports of increased as well as of decreased pump activity when the sex hormones were administered either in vivo or in vitro. Moreover, only rarely have isoform-specific or tissue-specific differences been addressed, and the results are inconclusive. Thus, much remains to be done.
In summary, the conclusions of Rathore et al. (32) concerning the role of gender in the outcome of digoxin therapy raise some interesting questions. Assessing for gender-related differences that may exist in clinical pathology and treatment of disease will, however, remain difficult unless greater numbers of women are enrolled in important clinical trials. Several seminal studies that have documented the life-saving benefits of angiotensin converting enzyme inhibitors (42) , aldosterone antagonism (43) , and β adrenergic receptor antagonism (44) have all been plagued by under-representation of female patients. Consequently, comparisons of therapeutic benefit have not had the same degree of statistical rigor in women as in men (32, 42–44) . Until such studies are designed to investigate potential gender-related differences, those therapies that have been documented to be beneficial in treating heart failure should be given to all CHF patients. Preclinical animal studies will also be needed to determine how gender and sex hormones affect the Na+ pump isoforms and the metabolism of cellular electrolytes in the heart and various other tissues. Such studies may provide clues to understanding gender-related differences in therapeutics.
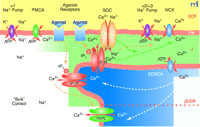
The plasma membrane-junctional sarcoplasmic/endoplasmic reticulum (PM-jS/ER) region (PLasmERosome). The important ion transporters involved in the local control of jS/ER Ca2+ stores and modulation of Ca2+ signals are shown. The PM overlying “bulk” cytosol contains Na+ pumps with α1 subunits (the “housekeeping” isoform that maintains the low, bulk cytosolic Na+ concentration) and PM Ca2+ pumps (PMCA). The PM microdomain overlying jS/ER contains Na+ pumps with α2 or α3 subunits, NCX, and store-operated channels (SOCs). α1 Na+ pumps are widely distributed in the PM but appear to be excluded from these microdomains. Agonist receptors may be expressed in these PM microdomains as well as elsewhere on the PM. The jS/ER contains S/ER Ca2+ pumps (SERCA), inositol trisphosphate receptors (IP3R) and ryanodine receptors (RYR). Although not illustrated here, the PM microdomain in some cell types (e.g., cardiac muscle and skeletal muscle) may contain L-type Ca2+ channels (dihydropyridine receptors), and the jS/ER may contain RYR. A very small, “diffusion-restricted” cytosolic space (J) lies between the PM and jS/ER; the concentrations of Na+ and Ca2+ may differ from those in bulk cytosol. Shading indicates relative concentrations of Na+ or Ca2+. The dotted orange line within the S/ER refers to the apparent compartmentalization of the S/ER in many types of cells. Only the upper compartment illustrated in the figure can be considered the junctional S/ER (jS/ER). ECF, extracellular fluid. Adapted from (45).
Acknowledgments
We thank Drs. Margaret M. McCarthy and Michael Fisher for helpful discussions.
- © American Society for Pharmacology and Experimental Theraputics 2003
References

Mordecai P. Blaustein, MD, (left front), is Professor and Chairman of the Department of Physiology and a Professor of Medicine. Shawn W. Robinson, MD, (right rear), is an Assistant Professor of Medicine (Cardiology Division) and of Physiology. Stephen S. Gottlieb, MD, (right front), is a Professor of Medicine (Cardiology Division). C. William Balke, MD, (center), is a Professor of Medicine and Head of the Division of Cardiology, and a Professor of Physiology. John M. Hamlyn, PhD, (left rear), is a Professor of Physiology. All are at the University of Maryland School of Medicine and are members of the University of Maryland Center for Heart, Hypertension, and Kidney Disease directed by Dr. Blaustein and co-directed by Dr. Balke. Address correspondence to MPB. E-mail mblauste{at}umaryland.edu; fax 410-706-8341.

