Adenosine Receptors and Mammalian Toll-Like Receptors: Synergism in Macrophages
- Mark E. Olah1 and
- Charles C. Caldwell2
- 1Department of Pharmacology and Cell Biophysics,
- 2Department of Surgery, College of Medicine, University of Cincinnati, Cincinnati, OH 45267
For many years, adenosine has had the reputation of being a “retaliatory metabolite” in that several actions of adenosine
are protective during periods of cellular stress (1). This designation was originally based in large part on the ability of adenosine to equalize energy supply to metabolic demand.
More recently, the role of adenosine as a protective signaling molecule has been explored extensively at both the physiological
and signal transduction levels in regard to its cardioprotective effects observed in cardiac preconditioning (2, 3), during ischemia–reperfusion injury (4), and in cerebral (5) and hepatic (6, 7) ischemia. During ischemic events, hypoxia promotes increased accumulation of adenosine to levels substantially greater than
those observed under normoxic conditions. Adenosine is able to produce its protective effects through activation of cell surface
G protein–coupled adenosine receptors (ARs) of which four subtypes are recognized, the A1 AR, A2A AR, A2B AR, and A3 AR (8).
The immune system represents another context in which the protective effects of adenosine are observed (9). Specifically, activation of the A2A AR inhibits several processes in polymorphonuclear leukocytes including degranulation (10), production of oxygen free radicals (11), and tumor necrosis factor–α (TNFα) (12), and adhesion to and migration across vascular endothelium (13, 14). Similarly, in macrophages, activation of the A2A AR decreases expression of TNFα and interleukin- (IL)-12 while increasing expression of the anti-inflammatory cytokine IL-10
(15). Adenosine may also modulate immune responses downstream of TNFα release, because adenosine suppresses TNFα-induced the activation
of the transcription factor nuclear factor–κ B (NF-κ B) in leukemic, lymphoid, and epithelial cells (16). Thus, in the immune system, adenosine may again act as a physiological “brake” in that, an unremitting inflammatory response
may result in organ damage or possibly lethal systemic inflammation. Indeed, the beneficial response to A2A AR agonists in ischemia–reperfusion injury may in large part reflect the anti-inflammatory effects of AR activation (4).
Two recent publications (17, 18) have demonstrated that activation of the A2A AR in murine macrophages produces, in addition to events typically deemed as anti-inflammatory such as downregulation of
TNFα, a regulation of vascular endothelial growth factor (VEGF). Treatment of macrophages with A2A AR agonists alone produces a relatively modest increase in VEGF secretion. However, in the presence of activators of mammalian
toll-like receptors (TLRs) that, by themselves, have negligible effects on VEGF expression, stimulation of the A2A AR results in increased VEGF secretion to a level similar to that produced under hypoxia, the best characterized and perhaps
most potent inducer of VEGF expression.
TLRs, of which at least ten family members exist, are critical components of the innate immune response. TLRs recognize and
are activated by foreign ligands such as lipopolysaccharides, a component of the outer membrane of Gram-negative bacteria,
bacterial DNA that contains unmethylated CpG dinucleotides, and flagellins (19, 20). Upon activation, TLRs convey a transmembrane signal(s) that ultimately results in the induction of pro-inflammatory cytokines,
and thus the immune response (19, 20). The role of the A2A AR in the observed synergy with TLRs to increase greatly VEGF expression was convincingly demonstrated through characterization
with AR subtype–selective agonists and antagonists, as well as through the examination of macrophages isolated from A2A AR-deficient mice (17). The dramatic effects observed upon coactivation of these two very different classes of receptors prompts speculation as
to the physiological importance of these interactions in the immune system and angiogenesis.
The ability of A2A AR agonists to inhibit TLR-induced TNFα production and yet, to act synergistically with TLR agonists to increase VEGF expression
led Pinhal-Enfield and coworkers (18) to suggest that these actions allow macrophages to switch from an inflammatory to an angiogenic phenotype. The term “angiogenic
switch” was introduced by Hanahan and Folkman (21) to describe of the events that promote vascular development in tumors. In this paradigm, either an increase in pro-angiogenic
factors or a decrease in anti-angiogenic factors, or both, creates an environment that promotes tumor growth through the development
of new vessels from the pre-existing vasculature (Figure 1⇓). A post-inflammation, angiogenic phenotype of macrophages would be of significance in wound healing, and indeed, activation
of the A2A AR is associated with enhanced wound healing in vivo (22).
Over the last decade, the most extensively examined pro-angiogenic molecule has been VEGF, a secreted protein that, through
activation of tyrosine kinase receptors, promotes key events in angiogenesis that include increases in vascular permeability,
and stimulation of endothelial cell proliferation and migration (23, 24). Interestingly, adenosine acting primarily through A2A AR or A2B AR also regulates endothelial cell function and promotes angiogenesis (25 –30). As with VEGF, adenosine- promoted angiogenesis may be considered beneficial in contexts such as wound healing (22) and myocardial ischemia (2), or detrimental in disease states such as cancer (31) or in retinopathy of prematurity (29). AR activation modulates VEGF expression; however, the direction of this regulation varies in an AR subtype–specific and
cell type–dependent fashion. For instance, activation of the A2A AR in macrophages stimulates VEGF expression (17,18), whereas this receptor subtype promotes VEGF downregulation in pheochromocytoma PC12 cells (32, 33). Stimulation of the A2B AR leads to increased VEGF expression in retinal endothelial cells (34), human microvascular endothelial cells (35), and human mast cells (30). Though the AR subtype was not precisely defined, an A2 AR has been implicated in increased VEGF expression in vascular smooth muscle cells (36). Future studies should further define the dependence of adenosine-induced endothelial cell responses (and subsequent angiogenesis)
on VEGF production. Additional AR subtypes may also be involved in regulation of angiogenesis through direct or indirect effects:
the activation of A3 AR results in increased expression of angiopoietin-2 in mast cells (30).
From the standpoint of the activation of receptors and their dowmsream effectors, the synergistic effect of A2A AR and TLR activation on VEGF production is provocative. Activation of TLRs does not increase A2A AR expression (17), thus the observed synergism is apparently not a simple amplification of the mechanism responsible for the relatively modest
increase of VEGF expression observed with A2A AR agonists alone. The A2A AR is classically associated with activation of the Gsα –adenylyl cyclase–cAMP-dependent protein kinase (PKA) cascade; however, the increase of VEGF in response to A2A AR and TLR activation is not sensitive to inhibition of PKA (17). The A2A AR may signal through G protein–coupled mechanisms that are not dependent on PKA activation (27, 33, 37). Thus, a possible role for these mechanisms (and even an absolute requirement for G protein–coupling) require future study.
The A2A AR–TLR synergism is not dependent on nitric oxide, which is required for the weak induction of VEGF by “TLR-only” agonists
(17). Based on the current data, it is tempting to speculate that simultaneous activation of TLRs and the A2A AR engages a signaling mechanism that is not stimulated by either receptor alone. TLRs, upon activation by ligand, act as
scaffolds with multiple signaling molecules including the MyD88 adaptor protein, the IL-1 receptor-associated kinase (IRAK),
and TNF receptor–associated kinase (TRAF6) recruited to the receptor (19, 20). Historically, G protein–coupled receptors were thought to be almost exclusively restricted to signaling by direct association
with G protein α subunits. More recently, however, G protein–coupled receptors are known to associate with additional proteins
involved in varied, but not necessarily mutually exclusive, processes including signal transduction, scaffolding, and cellular
localization (38, 39). Indeed, recent reports describe the physical association of the A2A AR with the glutamate mGlu5 receptor (40) and α-actinin (41). With strategies directed at detecting protein–protein interactions, such as yeast two-hybrid screening and resonance energy
transfer approaches, it may be possible to identify agonist-regulated association of the A2A AR and members of the TLR family. Because concomitant activation of A2A AR and TLRs result in increased VEGF mRNA expression (17), it may be hypothesized that the presently unidentified signaling cascade engaged by receptor coactivation ultimately regulates
VEGF gene transcription. However, regulation of VEGF expression may also occur through a modulation of transcript stability
(42).
In conclusion, apparently another facet of the protective effects of adenosine has been identified through the effects of
A2A AR activation on VEGF expression in macrophages with presumably important consequences in wound healing. Future studies should
reveal further information regarding the physiological significance of this response as well as reveal new signal transduction
mechanisms for both the A2A AR and TLRs.
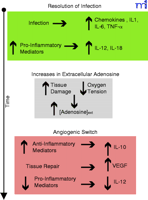
Figure 1.
Adenosine facilitates the angiogenic switch by macrophages. Upon bacterial infection, macrophages respond by the secretion of leukocyte recruiting mediators. Macrophages also secrete
cytokines that will initiate a Type 1 response by the adaptive immune system. This inflammatory response leads to increased
tissue damage and hypoxia as the bacterial infection is resolved (green). Extracellular adenosine concentrations increase
as a result of cellular damage and hypoxia (grey). Adenosine facilitates the angiogenic switch by inhibiting IL-12 and enhancing
VEGF and IL-10 production by macrophages (dark pink).
- © American Society for Pharmacology and Experimental Theraputics 2010
References
- ↵
Newby, A.C. Adenosine and the concept of “retaliatory metabolites.” Trends Biochem. Sci. 9, 42–44 (1984).
- ↵
Cohen, M.V., Baines, C.P., and Downey, J.M. Ischemic preconditioning: From adenosine receptor to KATP channel. Annu Rev. Phyiol. 62, 79–109 (2000).
- ↵
Sommerschild, H.T. and Kirkeboen, K.A. Adenosine and cardioprotection during ischaemia and reperfusion—an overview. Acta Anaesthesiol. Scand. 44, 1038–1055 (2000).
- ↵
Jordan, J.E., Zhao, Z.Q., Sato, H., Taft, S., and Vinten-Johansen, J. Adenosine A2 receptor activation attenuates reperfusion
injury by inhibiting neutrophil accumulation, superoxide generation and coronary endothelial adherence. J. Pharmacol. Exp. Ther. 280, 301–309 (1997). Using both an in vivo canine model of ischemia–reperfusion injury and in vitro assays of neutrophil activity, the authors
correlate A2AAR-mediated reduction of infarct size and inhibition of neutrophil function.
- ↵
von Lubitz, D.K.J.E. Adenosine and cerebral ischemia: Therapeutic future or death of a brave concept? Eur. J. Pharmacol. 371, 85–102 (1999). Review article that describes the protective effects of adenosine in cerebral ischemia. The complex roles of multiple AR subtypes
in neuroprotection as well as the clinical application of AR regulation in this therapeutic context are discussed.
- ↵
Carini, R., DeCesaris, M.G., Splendore, R., Vay, D., Domenicotti, C. Nitti, M.P., Paola, D., Pronzato, M.A., and Albano, E.
Signal pathway involved in the development of hypoxic preconditioning in rat hepatocytes. Hepatology 33, 131–139 (2001).
- ↵
Peralta, C., Closa D., Hotter, G., Gelpi, E. Bulbena, O., and Rosello-Catafau, J. Protective role of adenosine in inducing
nitric oxide synthesis in rat liver ischemia preconditioning is mediated by the activation of adenosine A2 receptors. Hepatology 29, 126–132 (1999).
- ↵
Fredholm, B.B., Ijzerman, A.P., Jacobson, K.A., Klotz, K.-N., and Linden, J. International Union of Pharmacology. XXV. Nomenclature
and classification of adenosine receptors. Pharmacol. Rev. 53, 527–552 (2001).
- ↵
Thiel, M., Caldwell, C.C., and Sitkovsky, M.V. The critical role of adenosine A2A receptors in downregulation of inflammation
and immunity in the pathogenesis of infectious disease. Microbes and Infection 5, 515–526 (2003). A recent and comprehensive review of the effects of A2AAR activation on multiple components of the immune system.
- ↵
Richter, J. Effect of adenosine analogues and cAMP-raising agents on TNF-, GM-CSF-, and chemotactic peptide-induced degranulation
in single adherent neutrophils. J. Leukocyte Biol. 51, 270–275 (1992).
- ↵
Cronstein, B.N., Rosenstein, E.D., Kramer, S.B., Weissmann, G., and Hirschhorn, R. Adenosine: A physiologic modulator of superoxide
anion generation by human neutrophils. Adenosine acts via an A2 receptor on human neutrophils. J. Immunol. 2, 1366–1371 (1985). Initial pharmacological demonstration that the inhibitory effect of adenosine on neutrophils is mediated specifically by the
A2AAR subtype.
- ↵
Thiel, M. and Chouker, A. Acting via A2 receptors, adenosine inhibits the production of tumor necrosis factor-α of endotoxin-stimulated
human polymorphonuclear leukocytes. J. Lab. Clin. Med. 124, 275–282 (1995).
- ↵
Cronstein, B.N., Levin, R.I., Philips, M., Hirschhorn, R., Abramson, S.B., and Weissmann, G. Neutrophil adherence to endothelium
is enhanced via adenosine A1 receptors and inhibited via adenosine A2 receptors. J. Immunol. 148, 2201–2206 (1992).
- ↵
Wakai, A., Wang, J.H., Winter, D.C., Street, J.T., O’Sullivan, R.G., and Redmond, H.P. Adenosine inhibits neutrophil vascular
endothelial growth factor release and transendothelial migration via A2B receptor activation. Shock 15, 297–301 (2001).
- ↵
Hasko, G., Szabo, C., Nemeth, Z.H., Kvetan, V., Pastores, S.M., and Vizi, E.S. Adenosine receptor agonists differentially
regulate IL-10, TNF-α, and nitric oxide production in RAW 2647 macrophages and in endotoxemic mice. J. Immunol. 157, 4634–4640 (1996). This report demonstrates the reciprocal regulatory effects of adenosine on macrophages through inhibition of IL-12 and TNFα
and the enhancement of IL-10 production.
- ↵
Majumdar, S. and Aggarwal, B.B. Adenosine suppresses activation of nuclear factor–κ B selectively induced by tumor necrosis
factor in different cell types. Oncogene 22, 1206–1218 (2003).
- ↵
Leibovich, S.J., Chen, J.-F., Pinhal-Enfield, G. et al. Synergistic upregulation of vascular endothelial growth factore expression
in murine macrophages by adenosine A2A receptor agonists and endotoxin. Am. J. Pathol. 160, 2231–2244 (2002). This paper and the one below are the focus of this Insight article. These reports describe the synergistic effects of A2AAR and toll-like receptor coactivation on VEGF production in macrophages.
- ↵
Pinhal-Enfield, G., Ramanathan, M., Hasko, G., Vogel, S.N., Salzman, A.L., Boons, G.-J., and Leibovich, S.J. An angiogenic
switch in macrophages involving synergy between toll-like receptors 2, 4, 7, and 9 and adenosine A2A receptors. Am. J. Pathol. 163, 711–721 (2003).
- ↵
Akira, S. Mammalian toll-like receptors. Curr. Opin. Immunol. 15, 5–11 (2003).
- ↵
Underhill, D.M. Toll-like receptors: Networking for success. Eur. J. Immunol. 33, 1767–1775 (2003).
- ↵
Hanahan, D. and Folkman, J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 86, 353–364 (1996). A review article that describes blood vessel development in tumors as related to a balance between pro-angiogenic factors
such as VEGF and inhibitors of angiogenesis such as angiostatin. Alteration of this balance results in the angiogenic switch
that promotes vascular sprouting.
- ↵
Montesinos, M.C., Desai, A., Chen, J.-F., Yee, H., Schwarzschild, M.A., Fink, J.S., and Cronstein, B.N. Adenosine promotes
wound healing and mediates angiogenesis in response to tissue injury via occupancy of A2A receptors. Am. J. Pathol. 160, 2009–2018 (2002).
- ↵
Zachary, I. and Gliki, G. Signaling transduction mechanisms mediating biological actions of the vascular endothelial growth
factor family. Cardiovasc. Res. 49, 568–581 (2001).
- ↵
Ferrara, N., Gerber, H.-P., and LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 9, 669–676 (2003).
- ↵
Meininger, C.J., Schelling, M.E., and Granger, H.J. Adenosine and hypoxia stimulate proliferation and migration of endothelial
cells. Am. J. Physiol. 255, H554–H562 (1988).
-
Meininger, C.J. and Granger, H.J. Mechanisms leading to adenosine-stimulated proliferation of microvascular endothelial cells.
Am. J. Physiol. 258, H198–H206 (1990).
- ↵
Sexl, V., Mancusi, G., Höller, C., Gloria-Maercker, E., Schütz, W., and Freissmuth, M. Stimulation of the mitogen-activated
protein kinase via the A2A-adenosine receptor in primary human endothelial cells. J. Biol. Chem. 272, 5792–5799 (1997). This paper, as well as additional reports from this group, explores the signaling mechanism that couples A2AAR activation to extracellular-signal regulated kinase stimulation and subsequent proliferation in endothelial cells. Results
indicate that A2AAR-mediated effects may not involve the classical Gsα–adenylyl cyclase–protein kinase A signaling cascade.
-
Dubey, R.K., Gillespie, D.G., and Jackson, E.K. A2B adenosine receptors stimulate growth of porcine and rat arterial endothelial
cells. Hypertension 39, 530–535 (2002).
- ↵
Afzal, A., Shaw, L.C., Caballero, S., Spoerri, P.E., Lewin, A.S., Zeng, D. Belardinelli, L., and Grant, M.B. Reduction in
preretinal neovascularization by ribozymes that cleave the A2B adenosine receptor mRNA. Circ. Res. 93, 500–506 (2003). A very recent publication that employs a novel technique to downregulate A2BAR expression to demonstrate the role of this receptor in endothelial cell function and angiogenesis in a murine model of
oxygen-induced retinopathy.
- ↵
Feoktistov, I., Ryzhov, S., Goldstein, A.E., and Biaggioni, I. Mast cell-mediated stimulation of angiogenesis: Cooperative
interaction between A2B and A3 adenosine receptors. Circ. Res. 92, 485–492 (2003). This paper reports that stimulation of ARs on mast cells promotes the release of angiogenic factors. In addition to stimulation
of VEGF expression by the A2BAR, activation of the A3AR promoted angiopoietin-2 expression.
- ↵
Spychala, J. Tumor-promoting functions of adenosine. Pharmacol. Therap. 87, 161–173 (2000).
- ↵
Kobayashi, S. and Millhorn, D.E. Stimulation of expression for the adenosine A2A receptor gene by hypoxia in PC12 cells: A
potential role in cell protection. J. Biol. Chem. 274, 20358–20365 (1999).
- ↵
Olah, M.E. and Gardner, A.M. Distinct protein kinase C isoforms mediate regulation of vascular endothelial growth factor expression
by A2A adenosine receptor activation and phorbol esters in pheochromocytoma PC12 cells. J. Biol. Chem. 278, 15421–15428 (2003). This paper and the one above describe the unexpected finding that activation of the A2AAR downregulates VEGF expression in PC12 pheochromocytoma cells. Adenosine generated during hypoxia may regulate VEGF expression
in a cell type–dependent manner.
- ↵
Grant, M.B., Tarnuzzer, R.W., Caballero, S., Ozeck, M.J., Davis, M.I., Spoerri, P.E., Feoktistov, I., Shryock, J.C., and Belardinelli,
L. Adenosine receptor activation induces vascular endothelial growth factor in human retinal endothelial cells. Circ. Res. 85, 699–707 (1999). This paper reports that activation of the A2BAR on human retinal endothelial cells stimulates both VEGF expression and cell proliferation. The importance of VEGF expression
in the A2BAR-induced cell proliferation is suggested by the finding that the response to an AR agonist is nearly abolished by VEGF antibody.
- ↵
Feoktistov, I., Goldstein, A.E., Ryzhov, S., Zeng, D., Belardinellia, L., Voyno-Yasenetskaya, T., and Biaggioni, I. Differential
expression of adenosine receptors in human endothelial cells: Role of A2B receptors in angiogenic factor regulation. Circ. Res. 90, 531–538 (2002). This report describes the differential expression of AR subtypes in human endothelial cells. Additionally, the A2AAR and A2BAR may have opposing effects on VEGF production.
- ↵
Gu, J.-W., Brady, A.L., Anand, V., Moore, M.C., Kelly, W.C., and Adair, T.H. Adenosine upregulates VEGF expression in cultured
myocardial vascular smooth muscle cells. Am. J. Physiol. 277, H595–H602 (1999).
- ↵
Liang, B.T. and Morley, J.F. A new cyclic AMP-independent, Gs-mediated stimulatory mechanism via the adenosine A2a receptor in the intact cardiac cell. J. Biol. Chem. 271, 18678–18685 (1996).
- ↵
Brady, A.E. and Limbird, L.E. G protein–coupled receptor interacting proteins: Emerging roles in localization and signal transduction.
Cell Signal. 14, 297–309 (2002).
- ↵
Hall, R.A. and Lefkowitz, R.J. Regulation of G protein–coupled receptor signaling by scaffold proteins. Circ. Res. 91, 672–680 (2002). A review article that describes interactions of GPCRs with various signaling proteins in G protein–dependent and –independent
contexts.
- ↵
Ferré, S., Karcz-Kubicha, M., Hope, B.T. et al. Synergistic interaction between adenosine A2A and glutamate mGlu5 receptors:
Implications for striatal neuronal function. Proc. Natl. Acad. Sci. U.S.A. 99, 11940–11945 (2002). Authors demonstrate the physical association of the A2AAR and mGlu5 glutamate receptor in transfected cells and in rat striatum that endogenously expresses the two receptors. In
addition to co-immunoprecipitation studies, functional consequences of receptor interactions are described.
- ↵
Burgueño, J., Blake, D.J., Benson, M.A. et al. The adenosine A2A receptor interacts with the actin-binding protein α-actinin. J. Biol. Chem. 278, 37545–37552 (2003).
- ↵
Iliopoulos, O., Levy, A.P., Jiang, C., Kaelin, W.F., and Goldberg, M.A. Negative regulation of hypoxia-inducible genes by
the von Hippel-Lindau protein. Proc. Natl. Acad. Sci. U.S.A. 93, 10595–10599 (1996).
-
Burgueño, J., Blake, D.J., Benson, M.A. et al. The adenosine A2A receptor interacts with the actin-binding protein a-actinin.
J. Biol. Chem. 278, 37545–37552 (2003).
-
Iliopoulos, O., Levy, A.P., Jiang, C., Kaelin, W.F., and Goldberg, M.A. Negative regulation of hypoxia-inducible genes by
the von Hippel-Lindau protein. Proc. Natl. Acad. Sci. U.S.A. 93, 10595–10599 (1996).
Mark E. Olah, PhD, (left) is Assistant Professor of Pharmacology and Cell Biophysics at the University of Cincinnati College of Medicine. His
research interest is signal transduction by adenosine receptors with particular focus on vascular endothelial growth factor
regulation and endothelial cell responses. Address correspondence to MEO. Email: Mark.Olah{at}UC.edu; fax 513-558-1169. Charles C. Caldwell, PhD, (right) is an Assistant Professor of Surgery in the Trauma, Sepsis and Inflammation Research Group at the University of
Cincinnati College of Medicine. He is currently studying the involvement of purinergic receptors and hypoxia inducible factors
on lymphocytes during trauma injuries and sepsis.