Protein KINASE C–Dependent Remodeling of Glutamate Transporter Function
Abstract
Glutamate is the predominant excitatory neurotransmitter in the mammalian central nervous system and is critical for essentially all physiological processes ranging from control of motor and somatosensory function to information processing and storage. Like many other small molecule neurotransmitters, transporters localized to the plasma membrane control the extracellular concentrations of glutamate. These transporters are both acutely and chronically regulated by several different mechanisms that presumably contribute to the protection of the nervous system from hypo- or hyper-glutamatergic function. In this review, we will describe our emerging understanding of one aspect of glutamate transporter regulation that is dependent on protein kinase C. More than a decade of extensive research on glutamate receptor–specific therapeutics has been driven by the hypothesis that these agents might be useful for pain management, treatment of schizophrenia or other psychiatric disorders, and prevention of neurodegenerative diseases. We assume that, in this modern era of drug discovery, understanding the endogenous regulatory mechanisms that are activated under physiological and pathological conditions will be required before one can target transporters for a ubiquitous neurotransmitter like glutamate.
Introduction
Glutamate is the predominant excitatory neurotransmitter in the CNS of vertebrates (1). Glutamate activates both ionotropic and metabotropic receptors (Figure 1⇓). Ionotropic receptors are ion channels mainly permeable to Na+, Ca2+, or K+, and are named for agonists that selectively activate the different subtypes, including N-methyl-d-aspartate (NMDA), α-amino-3-hydroxy-5-methylisoxazole-4-propionate (AMPA), or kainate. Metabotropic receptors are coupled to the activation of G proteins that subsequently regulate signaling pathways, such as adenylate cyclase or phospholipase C, and ion channels (2, 3). The total concentration of glutamate in the brain approaches 10 mmol/Kg (or 10 mM) and is 1000- to 10,000-fold higher than those of many other important neurotransmitters, including serotonin, norepinephrine, dopamine, and acetylcholine. Although several studies involving microdialysis suggest that extracellular concentrations of glutamate range from 1 to 10 μM (4, 5), these concentrations probably do not reflect the actual amount of glutamate in the synaptic cleft because low micromolar concentrations would tonically activate and/or desensitize receptors and mask synaptic signaling.
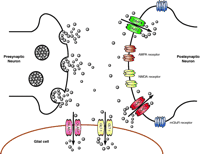
Schematic representation of a glutamatergic synapse. Synaptic release of glutamate results in activation of several different types of glutamate receptors that are situated on the post-synaptic neuron, on the presynaptic terminal, and the surrounding glial cells. For simplicity, receptors are only shown on the post-synaptic terminal. Glutamate is cleared from the synaptic space by transporters that are enriched on the postsynaptic termini and on the glial processes that sheath the synapse. Neurons primarily express the “neuronal” transporters EAAC1 and EAAT4, and astrocytes mainly express the “glial” transporters GLAST and GLT-1. The concentration of glutamate in the synaptic cleft has been estimated to be as high as 10 mM during periods of synaptic activation, but under resting conditions transporters probably reduce concentrations to less than 1 μ M.
There is no evidence for extracellular metabolism of glutamate; therefore, clearance by transporters is widely considered the main mechanism to control extracellular levels of glutamate. Two different transport systems have been identified. The first utilizes cation-derived electrochemical gradients to actively accumulate glutamate in both neurons and glial cells. These transporters are thought to function stoichiometrically to couple the inward movement of a molecule of glutamate to cotransport of three Na+ ions and one H+ (Figure 2A⇓). After the intracellular release of glutamate, Na+, and H+, a single K+ ion binds and is transported outward, facilitating reorientation of the transporter (6–8). The second transport system, called system Xc− is Cl− -dependent and is thought to mediate the exchange of cystine for glutamate across the plasma membrane (for discussion, see 9). It appears that there is no significant energy available to this system; therefore most investigators assume that this process has a passive, almost insignificant, role in the regulation of extracellular glutamate, although recent studies suggest that this assumption may not be correct (9).
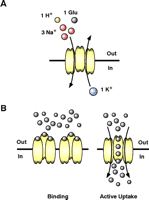
Transport through glutamate transporters. (A) Glutamate transporters use the Na+, H+, and K+ electrochemical gradients to drive the intracellular accumulation of glutamate into neurons and glial cells. Glutamate uptake is an electrogenic process that requires the inward movement of three Na+, one H+, and one molecule of glutamate coupled with the counter transport of one K+. (B) Glutamate transporters may reduce the amount of synaptic glutamate by two distinct mechanisms. The first mechanism involves glutamate binding that effectively buffers the amount glutamate available for receptor activation. Although this mechanism can be extremely fast, there needs to be a high density of transporters near glutamate release sites on astrocytes or neurons for this mechanism to be effective. The second mechanism involves active uptake. The cycle time for these transporters is estimated to be between 20 and 100 msec; therefore this process is slow compared to some of the very fast excitatory signals that can be a few msec in duration. However, under periods of sustained release or when the receptor signaling is relatively slow, active uptake can shape excitatory activity.
A Family of N a+ -Dependent Glutamate Transporters
Five different members of the Na+ -dependent transporter family have been identified, which include the L-glutamate–L-aspartate transporter [GLAST, also called excitatory amino-acid transporter 1 (EAAT1)], glutamate transporter 1 (GLT-1, also called EAAT2), excitatory amino-acid carrier 1 (EAAC1, also called EAAT3), EAAT4, and EAAT5. In addition, there is evidence for variants of these transporters that arise by alternate mRNA splicing (for original references, see 10–12). These different transporter subtypes share 40–65% amino-acid sequence similarity, but share no significant homology with the family of Na+ - and Cl− -dependent transporters that clear many of the other small molecule neurotransmitters, including γ -aminobutyric acid (GABA), glycine, dopamine, serotonin, and norepinephrine (11). Both the N and C termini of glutamate transporters are intracellular. Originally, hydrophobicity plots obtained from the predicted amino acid sequences of these transporters were interpreted to suggest between six to ten transmembrane domains (for reviews, see 10, 13). More recently, several groups have experimentally challenged these models and suggested that the C terminus may contain “pore loop” structures, similar to those observed in some ion channels, where the protein loops into and out of the membrane without spanning the membrane. In addition, one hydrophobic domain may traverse the surface of the membrane without actually entering the lipid bilayer (14–16).
The subtypes of glutamate transporters display differential patterns of distribution. Glial cells preferentially express GLT-1 and GLAST. GLT-1 is the predominant transporter in forebrain. Additionally, although it is expressed in astrocytes from most areas of the brain, GLT-1 is especially enriched in astrocytes from hippocampus and cortex. GLAST is mainly expressed in glial cells of cerebellum (17). EAAC1 and EAAT4 are considered neuronal transporters. EAAC1 is preferentially expressed in pyramidal cells from both hippocampus and cortex and EAAT4 is mostly expressed in Purkinje cells of the cerebellum (17, 18). Finally, the expression of EAAT5 is restricted to retina, and seems to be expressed both in glial cells and in neuronal cells (for review, see 10). Independent of the cell type, a common feature of glutamate transporters is that they seem to be enriched in membrane domains that face or surround the areas of glutamate release (12, 19).
Physiological and Pathological Functions of Glutamate Transporters
Several factors including the geometry of the synapse, the density and localization of the transporters, and the kinetics of receptor activation, inactivation, and desensitization influence both the amplitude and duration of excitatory signaling (for a review see 20). At synapses where there is a high density of glutamate transporters, the simple “binding” of glutamate to transporters may buffer the amount of “free” glutamate available to activate postsynaptic receptors (21). At other synapses, transporters appear to control the duration of receptor activation by active uptake (Figure 2B⇑) (22). Glutamate clearance by transporters has also been implicated in the modulation of synaptic plasticity. For example, in cerebellar synapses uptake by neuronal transporters modulates the activation of metabotropic receptors and the expression of long-term depression (LTD) (23). In area CA1 of the hippocampus, long-term potentiation (LTP) is associated with increases in glutamate uptake and increases in the cell surface expression of the neuronal transporter EAAC1 (24). Finally, in GLT-1 knockout mice, expression of LTP in hippocampus is impaired. This arises from chronic stimulation of glutamate receptors, because expression of LTP is recovered after a partial blockade of NMDA receptors (25). Thus, glutamate transporters likely participate in controlling receptor activation and synaptic plasticity.
Under physiologic conditions, uptake is so efficient that cells expressing glutamate receptors are quite resistant to direct glutamate injection; however, unabated extracellular accumulation of glutamate causes excessive activation of glutamate receptors (i.e., excitotoxicity) and cell death (26, 27). Different experimental strategies, including pharmacological inhibition, antisense knock-down or genetic deletion of the transporters, have been used to provide compelling evidence that decreased transporter function increases the vulnerability of neurons to excitotoxic insults (12, 28). Indeed, excitotoxicity contributes to the neuronal loss observed after acute insults to the CNS [e.g., stroke and head trauma (26, 27) ] and to the loss of oligodendroglia that occurs during hypoxic insults in the neonatal period (29). During these types of insults, the electrochemical gradients required to maintain active inward flux through the transporters collapse (30).
There is also evidence that excitotoxicity contributes to the loss of neurons observed in several chronic neurodegenerative diseases, such as Alzheimer Disease, amyotrophic lateral sclerosis (ALS), and Huntington Disease. In some cases, this may occur because of changes in transporter expression that accompany the progression of the disease. For example, in patients who die from ALS there is a dramatic reduction in expression of the GLT-1/EAAT2 subtype of transporter, and remarkably, this reduction selectively occurs in regions of the CNS in which the loss of neurons occurs (31). Similar changes in GLT-1 expression are also observed in animal models of this disorder (32).
It is entirely possible that different transporters will have different roles in excitotoxic processes observed in neurodegenerative diseases. For example, mice genetically deleted of the glial transporter, GLT-1/EAAT2, display dramatic reductions in brain glutamate uptake activity and develop lethal seizures similar to those caused by injection of glutamate receptor agonists that are not cleared by the transporters (33). In contrast, mice deleted of the neuronal transporter (EAAC1/EAAT3) show little change in total brain transport activity and no apparent signs of excitotoxicity (34) but show a delayed depolarization shift (a measure of glutamate release) after a hypoxic insult (35). This suggests that reversed operation of the neuronal transporter contributes to the rise in extracellular glutamate observed during a hypoxic insult. Therefore, if these transporters were differentially regulated, it might have a profound effect on how the nervous system responds to insults.
Glutamate transporter function seems to be tightly controlled at several levels, including transcriptional, translational, and posttranslational mechanisms. These transporters are regulated by a wide variety of substances including protein kinase activators, growth factors, and glutamate, itself. The levels of transport activity and transporter expression are altered in a number of pathological conditions such as hypoxia–ischemia, traumatic brain injury, epilepsy, and Alzheimer and Huntington diseases (10), implying that plasticity results in altered synaptic signaling and/or glutamate toxicity. In this review, we will focus on the regulation of glutamate transporters through the activation of protein kinase C (PKC).
Regulation of Transporter Activity
Early studies suggested that glutamate uptake may be regulated by a variety of signaling pathways (for reviews, see 11, 12). In most cases, these studies occurred before the different transporters were identified and before it was clear that multiple transporters can be expressed in the same tissue or cell; therefore, it was not possible to identify the specific transporter responsible for the observed changes (for reviews, see 11). One can also imagine that effects were missed because different transporters could be regulated in opposite directions by the same signaling pathway, with no net effect. In fact, in this review, we will describe evidence showing that PKC can have opposite effects on the glial transporter GLT1, and the neuronal transporter, EAAC1 (Figure 3⇓).
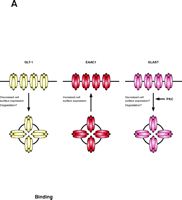
PKC activation differentially regulates the subtypes of glutamate transporters. Activation of PKC causes internalization of the glial glutamate transporter GLT-1, but increases the surface expression of the neuronal transporter EAAC1. One could imagine that simultaneous activation of PKC might provide a mechanism to shift clearance of glutamate from astrocytes to neurons. The activity of GLAST is also regulated by PKC. The specific mechanisms involved in regulation of the glutamate transporters have not been well explored but it is possible that they require the participation of “scaffolding” or “accessory” proteins to increase or reduce the number of active transporters located at the cell surface.
Advanced genetic techniques, including the molecular cloning of the different members of the glutamate transporter family, have allowed for the elucidation of the specific mechanisms involved in regulating each transporter subtype. Casado et al. demonstrated that phorbol ester–mediated activation of PKC rapidly increases the Vmax for glutamate uptake in astrocytes but not in neurons (36). Subsequently, this group suggested that GLT-1 is phosphorylated by PKC, and that elimination of a specific serine residue abolished the PKC-dependent increase in GLT-1–mediated activity (37). More recent studies have found different effects of PKC on GLT-1. For example, in human retinoblastoma cells that appear only to express the GLT-1 subtype, PKC activation causes decreased transport activity that is associated with an increase in transporter Km (38). We found that PKC activation did not change GLT-1–mediated uptake in two cell lines stably transfected with GLT-1 (39) or in the same cell line employed by Casado and colleagues. In contrast, using primary cultures derived from embryonic rat tissue, we found that activation of PKC causes a rapid (within minutes) decrease in cell-surface expression of GLT-1 without affecting total amounts of GLT-1, suggesting that PKC causes internalization of the transporter in this system (40). This effect is recapitulated in C6 glioma cells transiently or stably transfected to express GLT-1, and the decrease in cell-surface expression is associated with a decrease in transport activity that displays pharmacological properties consistent with GLT-1–dependent uptake. Using chimeric fusions of different transporter proteins, we identified a forty-three amino-acid domain located at the C terminus of GLT-1 as necessary and sufficient for PKC-dependent internalization of the transporter. Mutation of Ser486 within this domain partially abolished the PKC-induced redistribution of GLT-1, but did not reduce the phorbol ester–induced phosphorylation of GLT-1 (40). One possibility is that phosphorylation is unrelated and not required for regulated trafficking. A second possibility is that the amount of transporter phosphorylation is low, preventing detection either because the phosphorylated state is not stable or because it only represents a small fraction of the entire population. At present, it is not clear if the transporters internalized after PKC activation are targeted for degradation or recycled back to the plasma membrane. If both events can occur, this would provide alternative points at which transporter levels could be regulated. These varied effects of PKC activation on GLT-1 function suggest that additional proteins are required for regulated trafficking of GLT-1 and that these proteins are not uniformly expressed in different tissues, resulting in cell specific effects. Similar observations have been made for the insulin-dependent trafficking of the GLUT4 subtype of glucose transporter. This regulated trafficking is generally restricted to adipocytes and muscle cells; however, different mechanisms may be involved in these two cell types (41). We work with the assumption that primary cultures derived from brain tissue are more likely to possess a cellular milieu that more closely mimics that observed in vivo; therefore, we suspect that activation of PKC will cause an internalization of GLT-1 in vivo, but this has not been tested.
The regulation of GLAST by PKC has been examined in both heterologous and homologous systems. In Xenopus laevis oocytes expressing GLAST, activation of PKC reduces the currents that accompany the inward transport of glutamate. Although the reduction in glutamate uptake correlates with increased phosphorylation of GLAST, point mutations that eliminate the PKC-specific substrate consensus sites do not block the reduction in uptake nor do they block GLAST phosphorylation. This suggests that phosphorylation at consensus sites is not required for GLAST regulation and that the phosphorylation happens at nonconsensus sites (42). As determined by immunofluorescence, the reduction in uptake does not appear to be accompanied by a reduction in GLAST cell-surface expression, suggesting that the decrease in activity is not related to GLAST internalization (42). In cerebellar glial cells, activation of PKC reduces GLAST-mediated uptake. The decrease in transport activity correlates with a decrease in Vmax, suggesting that PKC may reduce the number of transporter molecules at the cell surface (43), but cell-surface expression has not been examined. In contrast, activation of PKC causes an increase in transporter Vmax in primary cultures of forebrain astrocytes (36, 44). GLAST is the only transporter detected in these culture preparations (45, 46). Curiously, preliminary studies from our laboratory show that activated PKC causes a loss of GLAST immunoreactivity, but the explanation for this effect is still unclear (47).
The above studies were undertaken by directly activating PKC with phorbol esters; however, some groups have begun to determine whether there are receptors that cause these changes in transport activity by a PKC-dependent pathway. One logical candidate would involve the activation of glutamate receptors. The presence of glutamate increases its own uptake in forebrain astrocytes and also increases the expression of GLAST at the cell surface, but these effects are not blocked by PKC antagonists (44). In Bergmann glia cells, glutamate reduces its own uptake through GLAST. However, in this case, PKC antagonists can block the glutamate-dependent decrease in GLAST-mediated transport activity, suggesting that this effect is PKC-dependent (48). In both systems, these effects appear to arise from the interaction of glutamate with the transporter itself. These studies suggest that in both forebrain and cerebellum, substrates provide a feedback mechanism to regulate transporter activity, but in opposite directions.
As is true for GLT-1 and GLAST, varied effects of PKC activation have also been observed for EAAC1. PKC activation rapidly increases EAAC1-mediated activity in C6 glioma cells, which endogenously express EAAC1. This increase in activity is associated with a redistribution of EAAC1 immunoreactivity from a subcellular compartment to the plasma membrane (49). Similarly, activation of PKC causes an increase in cell-surface expression of EAAC1 in neuron-enriched cultures (50). Do et al. found that activation of PKC also increases the currents associated with glutamate uptake in X. laevis oocytes expressing EAAC1 (51). In contrast, Trotti and colleagues reported that activation of PKC causes a decrease in EAAC1-mediated activity after expression in X. laevis oocytes or after stable transfection in Madin-Darby Canine Kidney cells (52). These effects were associated with a decrease in EAAC1 cell surface expression.
A number of studies have sought to determine which receptors might couple to PKC-mediated control of EAAC1. We have found that platelet-derived growth factor also increases EAAC1-mediated glutamate transport as well as EAAC1 expression at the cell surface, but these effects appear to be independent of PKC activation and may instead depend on activation of phosphatidylinositol 3-kinase (49). Neurotensin, a peptide with a variety of CNS effects, also increases both the activity and cell surface expression of EAAC1 in C6 glioma cells stably transfected with the high-affinity neurotensin receptor NTS1; however, this also seems to be independent of PKC or phosphatidylinositol 3-kinase activities (53). Thus, several different receptors appear to regulate EAAC1 redistribution and activity through different signaling molecules. Although no receptors have been shown to activate the PKC-mediated redistribution of EAAC1, inhalation anesthetics (e.g., isoflurane, halothane, and sevoflurane) increase EAAC1-mediated activity in C6 glioma by a PKC-dependent mechanism (51).
Multiple Subtypes of PKC: Possible Explanations for Differential Effects of PKC
As described above, PKC activation results in altered transporter activity and/or cell surface expression of the various glutamate transporters. In fact, several studies clearly document opposite effects of PKC activation on the activity of the same transporter expressed in different cellular systems, suggesting that cellular milieu and possibly different subtypes of PKC may explain some of these discrepancies. The PKC family is composed of three groups. The first group, termed classical PKCs includes three members (α, β, and γ) that are activated by diacylglycerol (DAG) and require Ca2+ as a cofactor. Members of the second group are referred to as novel PKCs and include four members (δ, ε, ??, and η). DAG activates these, but novel PKCs do not require Ca2+ as a cofactor. Both of these groups are activated by phorbol esters. The third group, known as atypical PKCs, includes two members (ζ and λ) that are insensitive to Ca2+, DAG, and phorbol esters (for reviews, see 54, 55). The biological importance of the different members of the PKC family has not been clarified. Some isozymes have overlapping expression patterns that may imply redundancy, expression of other subtypes is restricted to specific cell types, and some isoforms are restricted to subcellular organelles, suggesting that different subtypes may be involved in the regulation of different cellular functions.
Davis et al. found that the PKC-dependent increases in EAAC1-mediated glutamate transport were somewhat greater than could be explained by the increases in cell-surface expression of EAAC1 (49), suggesting that PKC may have two effects on EAAC1. We examined the subtypes of PKC that were expressed in C6 glioma cells in vitro and found that only PKCα (classical) and PKCε and PKCδ (both novel) were detected (50). Subsequently, using pharmacological agents to either selectively block or decrease expression of different PKC subtypes, we developed evidence that two PKC subtypes may be involved in the regulation of EAAC1. The first, PKCα, appears to mediate PKC-dependent increases in activity that are associated with an increase in EAAC1 surface expression. The second, PKCε, appears to increase EAAC1 activity by a mechanism that is independent of changes in the number of transporters at the plasma membrane, possibly by increasing the catalytic efficiency/turnover number of the transporters (50). More recently, we found that activation of PKC promotes an interaction between EAAC1 and PKCα. This interaction is specific for PKCα and is blocked by bisindolylmaleimide II (a general PKC inhibitor) and by Gö6976 (a classical PKC inhibitor) (56). This PKC-dependent interaction between EAAC1 and PKCα was observed both in C6 glioma cells and in crude synaptosomes prepared from rat brain.
In retinal Müller cells in situ, the general PKC inhibitor chelerythrine reduces GLAST-mediated uptake. This effect was mimicked by rottlerin (a putative specific inhibitor of PKCδ) but not by Gö6976 (an inhibitor of classical PKCs), suggesting that the baseline activity of GLAST is specifically maintained by the activation of PKCδ (57). Using cultured rat astrocytes our group has found that neither general PKC inhibitors (e.g., bisindolylmaleimide I and II, and staurosporine) nor Gö6976 had an effect on GLAST transport activity. However, within minutes of treating cells with concentrations of rottlerin similar to those required for inhibition of PKCδ, the capacity (Vmax) of GLAST to transport glutamate was decreased; this inhibition of activity was accompanied by a loss of GLAST immunoreactivity (58). Decreasing the expression of PKCδ did not block the rottlerin-induced inhibition of transport activity, suggesting that rottlerin regulates GLAST levels by controlling the rate of degradation or proteolysis through a target different than PKCδ (58). In contrast, in cultured Müller cells, activation of PKCδ with phorbol ester decreases glutamate uptake in a PKC-dependent manner (59). These apparently contradictory data may reflect differences in the target of rottlerin and the phorbol ester, or differences in the subtypes of PKC expressed in these two preparations, but this remains to be explored.
Finally, we have gathered preliminary evidence to suggest that Gö6976 but not rottlerin blocks the PKC-dependent internalization of GLT-1. We have also detected formation of complexes between GLT-1 and PKCα after PKC activation (60). These findings are similar to our previous observation linking the redistribution of EAAC1 to the activation of PKCα, and suggest that PKCα may also mediate the PKC-dependent internalization of GLT-1.
Many other members of the family of neurotransmitter transporters are also regulated by PKC activation, and in some cases there is evidence that specific PKC subtypes are involved in this regulation. In a recent report, Doolen and Zahniser show that the phorbol ester–dependent reduction in currents associated with dopamine transporter (DAT) activity are blocked by general PKC inhibitors, by an inhibitor of classical PKCs (Gö6076), by Ca2+ chelation (which should also be relatively selective for classical PKCs), and by an inhibitor of PKCδ (rottlerin). In contrast, a peptide that inhibits PKCε translocation and activation has no effect on the phorbol ester-induced inhibition of DAT currents. Based on these observations, the authors suggest that classical PKC(s) provide the primary mechanism for PKC-dependent regulation of DAT, and that the effect of rottlerin may be a nonspecific effect on the classical PKCs or other kinases (61).
These observations suggest that specific members of the PKC family are involved in the regulation of glutamate transporters, and furthermore, that general mechanisms may be in place to regulate the broader family of neurotransmitter transporters. If different PKC subtypes have specific or opposite effects on transporter trafficking and activity, this could also contribute to the heterogeneity of the responses observed in different cellular systems.
How Does PKC Change Transporter Function?
PKC-mediated increases or decreases in glutamate transporter function can be explained by either changing the number of transporters expressed at the cell membrane or by changing the intrinsic activity of the transporters already located at the cell surface. For some neurotransmitter transporters, the levels of cell-surface expression or the intrinsic activity can be regulated independently and by different mechanisms. It is possible that the formation of specific protein complexes may be involved in both types of regulation. Scaffolding proteins may facilitate the delivery or removal of transporter molecules to or from the cell surface and also may help to stabilize the transporters at the cell membrane. In addition, interactions with some accessory proteins may influence the catalytic efficiency of the transporter. In this section we will summarize the findings for some of the proteins that interact with neurotransmitter transporters.
Controlled protein trafficking is essential for regulating the activity of many different neurotransmitter transporters (for reviews, see 62–64). Delivery and removal of proteins to the cell surface may involve mechanisms that share similarity with other processes, such as the release of synaptic vesicles, implying that transporters exist on a defined compartment (i.e., transporter-containing vesicles) that allows the trafficking of transporter molecules to and from the plasma membrane. This possibility remains to be confirmed for glutamate transporters, but other members of the neurotransmitter transporter family––for example, the GAT-1 subtype of GABA transporter and the choline transporter––are found in vesicles. These vesicles contain some of the same constituents found in neurotransmitter-containing vesicles, including proteins required for fusion of these vesicles to the presynaptic terminal [e.g., soluble N -ethylmaleimide–sensitive attachment proteins (SNAPs) and SNAP receptors (SNAREs)] (65–67). Interfering with the function of one SNARE, syntaxin 1A––by anti-sense knockdown technology or proteolysis––modulates the cell-surface expression of GAT-1 and the norepinephrine transporter (NET) (68, 69). Furthermore, a physical interaction with syntaxin 1A may be required for the trafficking of GAT-1 and NET. PKC activation decreases the cell-surface expression of GAT-1 and increases the interaction of GAT-1 with syntaxin 1A (68). Thus, PKC might regulate the rate at which transporters are inserted in the plasma membrane by controlling the interaction between the transporter and syntaxin. The involvement of SNAREs in the regulation of glutamate transporters remains to be proven, but the intracellular expression of EAAC1, both in vivo and in vitro, is confined to discrete locations within the cell, suggesting the existence of a vesicular compartment available for redistribution to the cell membrane (12, 17, 49).
PDZ (post-synaptic density-95/Discs large/zona occludens-1) domain–containing proteins are involved in targeting, scaffolding, or clustering of a wide variety of membrane proteins (for review, see 70). In addition, they may serve as adapter proteins to promote a functional interaction between signaling molecules like PKC and a given transporter. One of these PDZ domain–containing proteins, Protein Interacting with the C-kinase (PICK1), specifically interacts with PKCα (71), and it is thought that PICK1 serves as an adapter to recruit PKCα into complexes to improve the specificity of PKC-dependent effects (72, 73). The C termini of DAT and NET each contain a PDZ binding domain that interacts with PICK1 (74). In both cases, the PICK1–transporter interaction appears to increase the number of transporters at the plasma membrane (74, 75). Although it is unclear how PICK1 increases the cell-surface expression of these transporters, it is possible that PICK1 anchors the transporters to the plasma membrane and decreases the rate at which they constitutively recycle between intracellular compartments and the plasma membrane. Because PICK1 interacts with the activated form of PKCα, it is possible that activation of PKC affects the stability of the transporters at the cell surface by regulating interactions between PICK1 and transporters. Preliminary evidence suggests that PICK1 interacts with one of the splice variants of GLT-1 (76) and with EAAC1 (77).
Activation of PKC phosphorylates and induces the rapid internalization of the serotonin transporter, leading to decreased transport capacity (78, 79). In a recent yeast two-hybrid screen, a homolog of the myristoylated alanine-rich C-kinase substrate (MARCKS) called MacMARCKS was isolated as a binding partner of the serotonin transporter (80). In coexpression experiments, MacMARCKS reduces the Vmax for serotonin uptake and blocks the PKC-induced loss of serotonin transporter from the cell surface (80). MARCKS proteins are phosphorylated in a domain enriched in basic amino-acid residues that are thought to interact electrostatically with the acidic lipids of the plasma membrane. In many cell types, phosphorylation by PKC abrogates the binding of MARCKS to the cell membrane (for review, see 81). These data are consistent with the idea that formation of MacMARCKS–serotonin transporter (SERT) complexes may be an intermediate step in the sequestration of SERT from the plasma membrane.
Modulation of the Intrinsic Activity
Previously we suggested that PKC regulates EAAC1 activity by a mechanism independent of transporter redistribution (49). Recently, we found evidence that PKC regulates EAAC1 catalytic efficiency and identified a non-classical PKC subtype, PKCε, as a candidate for this regulation. EAAC1 contains consensus sequences for PKC phosphorylation; thus, PKCε might directly phosphorylate EAAC1 and alter its intrinsic activity. However, PKCε might also increase the catalytic efficiency of EAAC1 by promoting the interaction of the transporter with an unidentified protein (50).
Although it is not clear how EAAC1 catalytic efficiency is regulated, studies of the regulation of GAT-1 catalytic efficiency by syntaxin 1A provide an example of how this might occur. GAT-1 regulation appears to involve at least three different proteins: PKC, syntaxin 1A, and munc18. Both syntaxin 1A and munc18 are proteins essential for intracellular membrane fusion. Under resting conditions, the availability of syntaxin 1A for interaction with GAT-1 may be limited by munc18. Activated PKC might then displace munc18 from syntaxin 1A–containing complexes, thereby increasing the number of syntaxin molecules available for interaction with GAT-1. The resulting increase in GAT-1–syntaxin 1A interactions causes a decrease in the catalytic activity of GAT-1 (68, 82, 83). Similarly, the interaction of syntaxin 1A with NET appears to reduce the intrinsic activity of NET, but it is not known whether PKC regulates this interaction (69). An alternative mechanism for the regulation of the catalytic efficiency of NET is observed in SK-N-SH cells. In these cells, insulin increases the activity of NET by a mechanism that appears to depend on phosphatidylinositol 3-kinase and p38 mitogen-activated protein kinase. This increase in NET activity is associated with an increase in transporter phosphorylation. NET activity and phosphorylation can also be regulated by blocking the activity of protein phosphatase 2A (PP2A), a protein known to interact with NET (84). Interestingly, PKC disrupts the interaction between NET and PP2A, suggesting that PKC may indirectly regulate NET phosphorylation and activity by modulating the NET–PP2A interaction (85).
Long-Term Regulation of Glutamate Transporters by PKC
All of the effects described in the previous sections occur within minutes and are likely to be independent of synthesis of new transporter molecules. Several studies have also shown that expression of glutamate transporters is regulated by a wide variety of signaling molecules, including growth factors, glutamate, and kinase activation (11, 12). These effects are likely to be related to alterations in transcription and translation or other mechanisms that change the total number of transporters. In many cases, these regulatory mechanisms also appear to depend on PKC. In astroglial cells, application of pituitary adenylate cyclase–activating peptide (PACAP) increases transport activity and expression of GLT-1 and GLAST. The PACAP-dependent induction of GLAST expression requires the activation of protein kinase A (PKA), whereas the PACAP-dependent induction of GLT-1 is dependent on the activation of PKA and PKC (86). Also in astrocytes, fibroblast growth factor, insulin-like growth factor, and epidermal growth factor increase GLAST mRNA and protein expression, but only the effects of insulin-like growth factor and epidermal growth factor are dependent on PKC. In Bergmann glia cells, long-term treatment with glutamate reduces GLAST mRNA and protein levels, and these effects are partially blocked by PKC antagonists (87). Direct activation of PKC using phorbol esters also reduces the level of GLAST mRNA and protein expression (43, 88). These observations suggest an important role of PKC in the regulation of GLAST expression, but the specific events involved in the regulation of expression of the glutamate transporters are still not well defined. The recent cloning of putative promoter elements for GLT-1 and GLAST will allow a more direct evaluation of the impact of the different signaling pathways on the regulation of the transcription of these transporters (88–90). The core of the GLAST promoter is apparently contained within a sequence of ~1.5 Kb, and a smaller fragment of ~0.7 Kb shows maximal reporter expression, suggesting the presence of both enhancer and repressor elements in the larger fragment (89, 91). Both phorbol esters or glutamate reduce reporter expression in constructs containing the GLAST promoter, suggesting that PKC can directly repress GLAST transcription. In this case, repression appears to depend on activation of the activator protein-1 (AP-1).
Summary
Glutamate transporters terminate the action of the main neurotransmitter in the mammalian brain. Recent studies have begun to define the mechanisms that regulate these transporters both acutely and chronically. Activation of PKC seems to be a ubiquitous mechanism for the regulation of the different members of the glutamate transporter family; however, the understanding of the molecular mechanisms involved in this regulation is only beginning to emerge. As these studies progress, it is becoming evident that glutamate transporters require a very complex arrangement of cellular events to function properly. The relevance of this regulation in normal and disease states is only beginning to be understood, but one assumes that understanding the regulatory mechanisms activated under physiologic and pathologic conditions will help to alleviate the adverse effects observed in many pathologies involving the disruption of glutamate metabolism.
Acknowledgments
Marco I. González is supported by a Postdoctoral Fellowship Award from the American Heart Association. Michael B. Robinson and the laboratory are supported by National Institutes of Health Grants (NS29868, NS36465, and NS39011).
- © American Society for Pharmacology and Experimental Theraputics 2004
References

Michael B. Robinson, PhD, is a Stokes Investigator at the Children’s Hospital of Philadelphia and a Research Associate Professor of Pediatrics and Pharmacology at the University of Pennsylvania. Address Correspondence to MBR. E-mail: robinson{at}pharm.med.upenn.edu; fax 215-590-3779.

Marco I. González, PhD, is a Postdoctoral Fellow in the Departments of Pharmacology and Pediatrics at the University of Pennsylvania and the Children’s Hospital of Philadelphia.

