Allosteric Modulators: The New Generation of Receptor Antagonist
Abstract
Allosteric antagonists modulate the affinity and/or efficacy of agonists for receptors. Although the manner in which this modulation can occur can mimic that of simple competitive antagonists, allosteric antagonists possess unique properties that can present seemingly capricious profiles of antagonism. These unique properties also offer potentially useful patterns for therapeutic utility. This review summarizes methods to detect allosteric antagonism and some special properties of these receptor modulators.

Introduction
In the pharmacological lexicon, drugs generally are classified as either agonists or antagonists in any given system. In the case of agonists, it is common to expect complex system interactions that involve the interplay of the molecular properties of affinity and intrinsic efficacy; thus, agonists can be potent but not efficacious, resulting in a level of responsiveness far below the maximal level, or they can be efficacious but not potent (Figure 1⇓). In contrast, antagonists have been presented as being more simple, with activity conceputalized as a reflection of affinity alone. Thus, in an orthosteric context, where an antagonist binds to a site required by an agonist, equiactive concentrations of antagonists (i.e., concentration of a given antagonist expressed relative to its equilibrium dissociation constant) will provide equivalent amounts of antagonist-blocked receptor species. Thus, there is no “pharmacological texture” with respect to which orthosteric antagonist is chosen to block an effect; antagonists are equivalent. Such simplicity is inconsistent within the allosteric context, where antagonists can bind simultaneously with the agonist and thus can selectively permit signals to be propagated through the receptor. In these cases, all allosteric antagonists are not equivalent, and a great deal of “texture” with respect to the antagonist-blocked receptor can result.
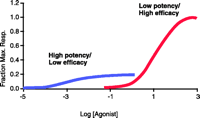
Affinity and efficacy. Two types of agonist reflect differences in affinity and efficacy; an agonist can be affinity dominant (high affinity, low efficacy) or efficacy dominant (high efficacy and low affinity). Together, the two properties define the agonist potency profiles.
The classical model of allosteric antagonism, as presented by Ehlert (1), is shown in Box 1. According to this model, the fraction (ρ) of receptor species bound by agonist A (eliciting a pharmacological response) in the presence of allosteric antagonist/ modulator B can be expressed as:
The Classical (Ehlert) Model For Receptor Allosterism
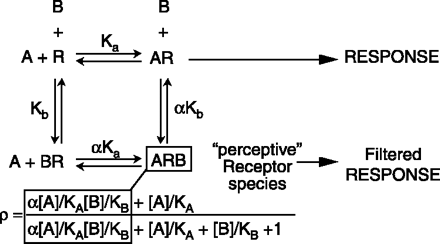
The receptor binds a tracer ligand, or probe, (agonist A) and an allosteric antagonist molecule (denoted B) with respective affinities Ka and Kb. When B is bound the receptor can still respond to A; the degree of this response depends on the nature of A (i.e., probe dependence). When one of the ligands is bound, the affinity of the other ligand is modified by a factor denoted αand referred to as the cooperativity constant. If α>1, then the binding is “positively cooperative,” which signifies that the affinity of A is higher for the receptor when B is bound and vice versa; B can be said in this case to mediate “allosteric potentiation.” If α<1, then the binding is antagonistic; the affinity of A is lower for the receptor when B is bound. Model presented by Ehlert (1). Direct evidence for the existence of a “perceptive” receptor species has been shown in the case of the CC chemokine receptor 5 (CCR5) through the use of the allosteric antagonist 873140 and the probe ligand RANTES (2). The terms for the “perceptive” receptor species ARB are in the white squares for the equation defining response in an allosteric system.
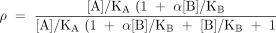
where KA and KB are the respective equilibrium dissociation constants of the ligand–receptor complexes) and αis a cooperativity constant (see Box 1). This equation predicts a prominent and unique feature of allosteric antagonists, namely, that their effects are saturable, which is readily seen from the instance where the concentration of antagonist approaches infinity. Specifically, in the absence of antagonist (i.e., [B]/KB=0) the location parameter of the agonist dose-response curve is represented, by definition, as KA; in the presence of antagonist, this location parameter is modified to become: KA([B]/KB+1)/(1+α[B]/KB). Thus, the agonist dose ratio is given by:
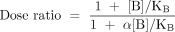
It is thus apparent that as the concentration of antagonist B approaches infinity, the maximal dose ratio is finite and approaches α−1. This finite limit is in contrast to an orthosteric simple competitive antagonist, where the dose ratio for an agonist in the presence of an infinite concentration of antagonist also is infinite.
The second property of allosteric antagonism is illustrated in the Figure for Box 1. Specifically, it can be seen that a receptor species exists that binds both the agonist and antagonist concomitantly. This receptor species is said to be “perceptive,” in that it reacts to both agonist and antagonist and perceives the presence of each. The perceptive receptor species theoretically allows a portion, or even all, of the agonist stimulus to be passed on to the system. Thus, the binding of an allosteric antagonist may be catastrophic for one agonist and inconsequential for another; the antagonism is probe dependent.
The saturability and probe dependence of allosteric antagonism gives a texture to allosteric antagonists much like that existing for agonists. The cooperativity factor α can be thought of as an indicatior of “efficacy” of an allosteric antagonist; the value of α that describes antagonist efficacy will vary for each agonist. An appreciation of these properties is essential to an understanding of the effects of allosteric antagonists in receptor systems and will allow us to exploit their potential therapeutic relevance.
Mechanism of Receptor Allosterism
Allosteric antagonists produce unique effects by binding to a site on the receptor to produce a bias in the receptor conformation. These effects can be remarkable, in some instances changing the order of potency for a series of agonists. Such a reordering of potency has been observed for muscarinic M2 receptors, where the unmodulated order of agonist potency [i.e., Arecaidine propargyl ester (APE)> Arecoline > Oxotremorine > Methylfurmethide = Pilocarpine >Acetylcholine > Carbachol] is, in the presence of the allosteric modulator eburnamonine, drastically rearranged, to become: Arecoline > APE > Methylfurmethide > Oxotremorine > Carbachol> Acetylcholine > Pilocarpine (Figure 2⇓) (3). In terms of classical receptor pharmacology, such perturbation of the rank order of agonist potency is indicative of distinct receptor subtypes, and in consideration of allosteric effect, these data highlight an important concept. Specifically, an allosterically modulated receptor can in some ways be thought of as a distinct receptor subtype relative to the native (unmodulated) receptor, having its own characteristic set of affinities.
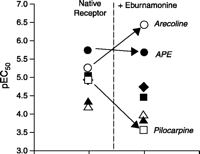
Allosteric modulation of receptor responses to a series of agonists. Potency values [expressed here as pEC50 values (i.e., the negative logarithm of the molar concentration producing the half-maximal response) of muscarinic agonists for M2 receptors are indicated in the absence and presence of saturating concentrations of the allosteric modulator eburnamonine. Acetylcholine (Ach, filled triangles), pilocarpine (open squares), oxotremorine (filled squares), methylfurmethide (filled diamonds), arecaidine propargyl ester (APE, filled circles), arecoline (open circles) and carbachol (open triangles). Data from (3).
Although allosterism imparts a change in receptor shape that results in a different responsiveness to ligands, it is not inevitable that all shape changes will result in antagonism. Thus, the term “antagonist” can be ambiguous (if not misleading) in the allosteric context; the line separating antagonism and potentiation is blurred and probe dependent. For example, N-chloromethylbrucine increases the affinity of muscarinic M3 receptors for acetylcholine (4), and the allosteric ligand alcuronium completely blocks the binding of methyl quinuclidinyl benzilate but elicits a 250% increase in the binding of atropine (5). As seen in Figure 2⇑, eburnamonine is an antagonist of pilocarpine at the M2 receptor, is neutral with respect to APE agonism, and potentiates arecoline agonism. For this reason, it may be more appropriate to refer to these ligands as “modulators,” as this term suggests alteration of initial strength of signal without connoting the quality of modulation (e.g., antagonism or potentiation).
The scale of allosteric effects, in terms of the size of molecules affected, can vary greatly. Figure 3⇓ shows allosteric antagonists of the chemokine receptor CCR5, which vary from the small CCR5 antagonist 873140 (0.6 kDa) to the large envelope protein (gp120) of the human immunodeficiency virus (HIV). Interestingly, blockade of HIV entry is effected by 873140 at nanomolar concentrations, yet it is postulated that large areas of the CCR5 receptor and gp120 interact to promote HIV entry into cells [see (6) for review]. Thus, it is unlikely that single-point steric hindrance mediated by binding of 873140 is responsible for its potent inhibition of HIV entry. So, how can a molecule 0.5% of the size of the proteins involved prevent a tight binding interaction between them? One possible mechanism is that a global change of the receptor conformation occurs with the binding of 873140, which perturbs numerous points of interaction between the receptor and gp120. Thermodynamic considerations also suggest a basis for the inhibition, namely, a mechanism of conformational selection (7). It is known that proteins adopt numerous conformations in pharmacological space in response to changes in thermal energy (8–13). Ligands with selective affinity for a limited number of these conformations can lead to a change of the ensemble of conformations available to react with other proteins or ligands (14 ,15). If the binding of 873140 favors conformations that do not support HIV infection, then this ligand will be therapeutic irrespective of the size of the proteins involved.
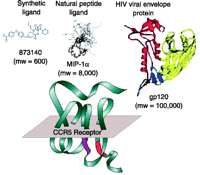
Relative sizes of high-affinity ligands of the CCR5 chemokine receptor. 873140 is a synthetic small-molecule (0.6 kDa) non-competitive allosteric antagonist. MIP-1α(8 kDa) is a natural peptide ligand for the receptor. The viral envelope protein gp120 (120 kDa) mediates the interaction between CCR5 and HIV.
Another prediction from the mechanism of conformational selection is that the allosterically ”selected” (i.e., blocked) CCR5 conformations need not be similar in any way other than the fact that they do not support HIV binding. Because the conformational changes in protein structure they induce can be global, allosteric antagonists may prove to be powerful in regards to thwarting viral resistance to drugs. Changes in HIV envelope protein, for example, have been shown to alter viral tropism, that is, to change the receptor used for HIV entry from CCR5 to CXCR4 (16–19). It is theorized that chronic treatment with one CCR5 antagonist may provide selective pressure under which the virus becomes better suited for survival—in effect, HIV can “learn” to re-configure the envelope protein, through mutation, so that it can overcome certain therapies (20–23). Experimentally, passage studies with AD101, an antagonist structurally related to the allosteric antagonist Sch-C, have shown resistance through the emergence of an escape mutant in the presence of antagonist (24). It may thus be argued that blockade of CCR5-mediated HIV entry would best be implemented through different allosteric antagonists that would produce a novel ensemble of CCR5 conformations, thereby rendering HIV mutants unable to interact and proceed with infection. In this way, successive use of different allosteric modulators may be an important approach to overcoming viral resistance.
The probe dependence of allosteric antagonism also has broad implications for therapeutic intervention, providing an obvious guideline for discovery of allosteric antagonists. Specifically, screening and lead optimization of allosteric antagonists must be done with the physiologically relevant receptor probe (e.g., ago-nist). For example, to screen potential drugs that might potentiate cholinergic function in Alzheimer’s disease, a ligand-binding assay might theoretically be used to detect allosteric perturbation of acetylcholine receptors in their ability to bind [3H]N-methylscopolamine (NMS). The use of NMS as a probe, however, may not accurately reflect the way that allosteric perturbations might alter the binding of acetylcholine, the physiological ligand. Indeed, strychnine produces a 2.4-fold potentiation of receptor affinity for [3H]NMS, suggesting perhaps that strychnine-based chemical derivatives should be pursued for further structure–activity relationships; however, strychnine produces tenfold antagonism of the natural ligand acetylcholine (25). Thus, in this case, the incorrect choice of probe molecule would yield misleading results with respect to potential therapeutic value.
The Geography of Allosterism
By definition, an allosteric receptor protein will bind the molecule (either agonist or radioligand) used to probe receptor function at a site separate from that utilized by the allosteric modulator, and any degree of cooperativity between these two sites (i.e., potentiation or antagonism; see Box 1) will be mediated through the protein itself. Allosterism thus has three important ramifications. The first, as discussed previously, is saturation of effect (i.e., potentiation or antagonism), which indicates that the binding site recognized by the potentiator/antagonist is fully occupied. Increases in concentration beyond those required for essentially maximal receptor occupation therefore will not increase the magnitude of the allosteric effect; note, however, that increases in the duration of effect can nevertheless result. This separation of magnitude of effect from duration of effect is in contrast to orthosteric antagonists, where increases in concentration are inexorably linked to increases in maximal effect.
The second implication of allosterism relates to selectivity of effect. For example, the muscarinic receptor subtypes M1 to M5 all must recognize acetylcholine; however, allosteric sites on these receptor subtypes may (or may not) be selective for distinct modulator molecules. (Orthosteric antagonists, on the other hand— binding to the same site as acetylcholine—would be far less likely to be specific for a given subtype.) Accessory allosteric sites on these receptors, furthermore, may be heterogeneous, thus offering an additional dimension of selectivity (26). Table 1⇓ shows the greater range in potency of four allosteric antagonists for muscarinic receptor subtypes (100-fold) as compared to muscarinic agonists (3). On the other hand, an allosteric recognition site can be common not only among receptor subtypes, but even across distinct receptors. For example, the receptor antagonist SCH202676 has micromolar potency for opioid, dopamine, adrenergic, and muscarinic receptors (Figure 4⇓) (27).
Selectivity of Agonists and Allosteric Antagonists for Muscarinic Receptor Subtypes: pK A 1 or pK B 2 values
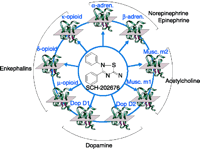
Interaction of the allosteric ligand SCH-202676 with multiple receptor types. The allosteric ligand SCH 202676 binds to an allosteric site on dopamine D1, D2, opioid μ, α, κ, muscarinic M1, M2 receptors, and α- and β-adrenoceptors. After (27).
The third implication of separate binding sites involves the permissive nature of allosteric antagonism. Specifically, the existence of perceptive receptor species (Box 1) allows the allosterically modulated receptor to respond to agonist in a manner dictated by the cooperativity constant α. Thus, any pattern of physiological tone imparted by endogenous agonist can be modulated by an allosteric antagonist. Such modulation may be therapeutically relevant in cases such as Alzheimer’s disease, where augmentation of failing cholinergic function could be useful (28–32). In such cases, complex central neural patterns would be preserved by an allosteric ligand that could potentiate cholinergic receptor function (α>1). Such potentiation, of course, would not be a feature of general orthosteric cholinergic agonism.
Masquerading Antagonists
The characteristics that distinguish allosteric ligands from standard orthosteric competitive antagonists are summarized in Table 2⇓. Having discussed the importance of differentiating between orthosteric and allosteric antagonism (in order to understand full therapeutic utility), we can now consider what pharmacological tools are available to do so. The main characteristics we will look for are antagonist saturability and probe dependence. Figure 5⇓ shows common behaviors of orthosteric and allosteric antagonists. With appropriate values of α and within a limited range of concentration, an allosteric antagonist could well masquerade as a simple competitive antagonist by producing parallel shifts (to the right) in agonist dose-response curves and maintaining maximum response and sigmoidal antagonist displacement curves (in radioligand binding). To expose such a masquerade, extension of the normal quantitative studies utilized to measure antagonism can differentiate allosteric from orthosteric antagonism. For example, a simple competitive antagonist offering mere steric hindrance to agonists will yield the same degree of antagonism regardless of which agonist is used as a probe (reflected by a linear Schild regression with identical pKB values). In contrast, different agonists may uncover differing degrees of antagonism (different values of α) in accordance with the permissive nature of allosteric antagonism (see the effects of eburnamonine at the M2 receptor discussed above). Similarly, as shown in Figure 6⇓, allosteric antagonists, which are saturable, can in some cases fail to fully displace radiolabeled probes of agonist binding; this property can thus be diagnostic of allosteric antagonism.
Unique Features of Allosteric Ligands

Masquerading antagonists. Both allosteric and orthosteric antagonists can manifest identical behaviors, such as parallel shifts of dose-response curves (with no diminution of maxima) for simple competitive antagonists, depression of maximal response with no shift to the right for noncompetitive antagonists, and sigmoid binding curves (with complete displacement of radioligand). However, the behavior of these types of antagonist diverges in that, unlike orthosteric antagonism, allosteric antagonism can be saturable and probe dependent. Thus, simple competitive antagonists produce identical blockade of all agonists, yielding identical linear Schild regressions, and they also manifest complete displacement of probe radioligand with a linear relationship between the concentration of radioligand and IC50 for antagonism. In contrast, the maximal degree of functional antagonism by allosteric ligands may differ for different agonists, and their Schild regressions can be curvilinear and agonist dependent; radioligand displacement may be incomplete as well. (nsb, nonspecific binding)
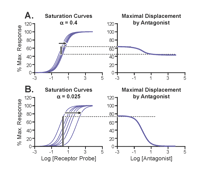
Displacement of radioligand by allosteric antagonists. (A) An allosteric antagonist with α= 0.4 produces a low-magnitude shift to the right of the saturation binding curve; correspondingly, a shallow curve is observed for displacement of 60% maximally bound radioligand. In contrast (B), allosteric antagonists with lower values of α (here, α= 0.025) and orthosteric antagonists produce much larger shifts to the right of the saturation binding curve, resulting in a complete displacement of pre-bound radioligand.
Allosteric antagonism, it should be noted, can be assay dependent. Box 2 shows a model of allosteric function extended from the Ehlert model by Hall (33) to include receptor activation. The Hall model predicts that receptor binding and functional assays can yield different degrees of apparent antagonism. For example, clopropan[b]chromen-1a-carboxylic acid ester produces non-competitive blockade of glutamate receptors in functional assays but no comcomitant effects on glutamate binding (Figure 7⇓) (34).
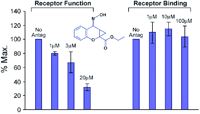
Receptor functionality vs receptor binding. The non-competitive antagonist clopropan[b]chromen-1a-carboxylic acid ester inhibits the function of a glutamate receptor without appreciably affecting glutamate binding. Data redrawn from (34).
When Binding and Function Don’t Agree
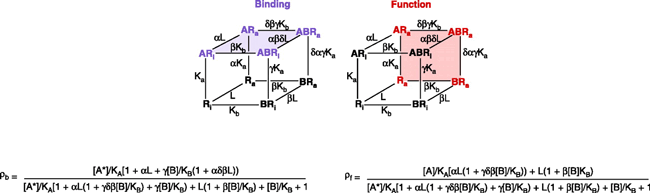
A model by Hall (33) describes the allosteric interaction between an agonist A (probe) and allosteric antagonist B with a receptor that can exist in an active (Ra) and inactive state (Ri). Binding assays measure the receptor species bound by the radioprobe A with no regard to the activation state of the receptor (blue receptor species). The equation for the amount of bound radioactive receptor species is shown under the schematic. In contrast, receptor function measures the amount of active state receptor species (Ra) and not necessarily the probe-bound species (receptor species in red). Under these circumstances, the amount of agonist response in the presence of an allosteric antagonist is given by the equation below the schematic; note the difference from the equation depicting receptor binding. Because of these differences, the two assays may return different indications of antagonist activity. It should be noted that the Ehlert model (1) can describe no effect on binding (α= 1) but inhibition of function if it is assumed that receptor species bound by allosteric modulator (i.e., BR and ARB) do not produce response.
Conclusion
Simple competitive antagonists can have texture related to conventional receptor intrinsic efficacy, that is, they can be partial or inverse agonists. However, allosteric antagonists have an added parameter describing molecular activity, namely, the cooperativity constant α. This constant also constitutes a type of efficacy (as described by modifying receptor behavior), which must be considered in investigations of receptor antagonists. The use of a variety of receptor probes (i.e., agonists or radioligands) is a powerful tool in the identification of allosteric antagonism; antagonists can be potent blockers of one agonist and relatively inert with respect to another. The methodologies that identify allosteric antagonism will need to be increasingly considered as industrial screening continues to move toward functional assays of antagonism (as opposed to binding studies) (35). The unique properties of allosteric antagonists clearly set them apart from standard competitive antagonists; tools for exploring these properties exist and may offer the means for exploiting the untapped potential of permissive antagonists.
- © American Society for Pharmacology and Experimental Theraputics 2004
References

Terry Kenakin, PhD, studies and applies concepts of receptor pharmacology to drug discovery programs at GlaxoSmithKline Research in Research Triangle Park, North Carolina. He is interested in receptor mechanisms and the system-independent quantification of drug activity. E-mail Terry.P.Kenakin{at}gsk.com; fax 919-483-0585.

