MOLECULAR EFFECTS of lithium
Abstract
Bipolar affective disorder is a common, severe, chronic, and often life-threatening illness, associated with other medical and psychiatric conditions (i.e., co-morbidity). The treatment of this devastating disorder was revolutionized by the discovery of lithium’s antimanic effects over fifty years ago. Recent molecular and cellular biological studies have identified a number of unexpected targets for this monovalent cation, notably glycogen synthase kinase-3 and neurotrophic signaling cascades. These findings are leading to a reconceptualization of the biological underpinnings of bipolar disorder and are resulting in considerable interest in utilizing lithium for the treatment of certain neurodegenerative disorders. We review recent insights into lithium’s actions including its direct inhibitory actions on inositol monophosphatase, inositol polyphosphate 1-phosphatase, glycogen synthase kinase-3, fructose 1,6-bisphosphatase, bisphosphate nucleotidase, and phosphoglucomutase enzymes. We also discuss lithium’s intracellular downstream targets including adenylate cyclase, the phosphoinositol cascade (and its effect on protein kinase C), arachidonic acid metabolism, and effects on neurotrophic cascades. Many of the new insights of lithium’s actions may lead to the strategic development of improved therapeutics for the treatment of bipolar disorder.

Introduction
Bipolar disorder is a devastating and relatively common disease, with an overall lifetime incidence of about 1% in the general population. A number of studies show that for a high percentage of patients the outcome is poor, with a high rate of chronicity, residual symptoms, relapse, subsyndromes, cognitive and functional impairment, and psychosocial disability (1, 2). The costs associated with disability and premature death represent an economic burden of tens of billions of dollars annually in the United States alone; not surprisingly, the Global Burden of Disease Study has identified bipolar disorder and mood disorders among the leading causes of disability worldwide, with increasing disability likely in the coming years (3). In addition to the tremendous economic cost, suicide is estimated to be the cause of death in 10–20% of the individuals with bipolar disorder, and increasingly, mood disorders are associated with many other health-related consequences (4, 5).
The discovery of lithium’s efficacy as an antimanic agent over fifty years ago revolutionized the treatment of patients with bipolar disorder. The remarkable efficacy of lithium has served to spark a revolution that has, over time, reshaped not only medical and scientific but also popular concepts of severe mental illnesses. Indeed, the efforts to understand how a simple monovalent cation like lithium can exert such profound beneficial effects has led investigators to examine the signal transduction pathways involved in bipolar disorder. After nearly fifty years, lithium continues to be one of the mainstays of treatment for this disorder, both for the acute manic phase and as prophylaxis for recurrent manic and depressive episodes. Adequate lithium treatment, particularly in the context of a lithium clinic (an outpatient clinic dedicated to the treatment of bipolar patients and psychopharmacological management of lithium), also reportedly reduces the excessive mortality observed in the illness (6, 7). In the last decade, there has been an explosion in the number of options available for the treatment of recurrent mood disorders with a parallel and unprecedented increase in the interest in the treatment of bipolar disorder by pharmaceutical companies, clinicians, researchers, and indeed the general public. Despite the introduction of a number of new anticonvulsants and antipsychotics into the pharmacopeia, the last three years have seen a resurgence of interest not only in lithium’s utility in the long-term treatment of bipolar disorder, but possibly also for neurodegenerative disorders. This renewed interest in lithium has come about largely due to converging evidence from biochemical studies that have identified critical signaling and neurotrophic molecules as targets for lithium’s actions.
In spite of lithium’s past success and future promises, however, it remains far from the perfect drug. Increasing evidence suggests that a significant number of patients do not respond adequately or cannot tolerate its side effects, or both. Similarly, other mood stabilizers such as valproate (VPA) and carbamazepine are ineffective or intolerable for a significant proportion of patients. The recognition of the significant morbidity and mortality of patients with severe mood disorders as well as the growing appreciation that a significant percentage of patients respond poorly to existing treatments have made the task of discovering new therapeutic agents that are both efficacious and have few side effects increasingly more important. We discuss these recent insights into lithium’s actions and discuss their implications not only for changing our existing concepts of the pathophysiology of severe mood disorders, but also for the strategic development of therapeutics that possess better tolerability, fewer side effects, and better toxicity profiles and pharmacodynamic characteristics than lithium, which may improve the treatment of bipolar disorder. It is hoped that by understanding lithium’s true therapeutic target we will be able using a hypothesis driven approach, to attempt to treat patients with novel drugs with lithium-mimetic properties. We begin with an overview of lithium’s direct targets and follow with a discussion of adaptive changes, which are observed with chronic lithium administration in a therapeutically relevant time frame.
Direct Targets of Lithium
Lithium has an ionic radius that is similar to that of magnesium, and inhibits some enzymes through competition for this often required cofactor (8–10) (Table 1⇓). Although lithium inhibits, to some degree, a number of enzymes (11), only a few enzymes are significantly inhibited at therapeutic serum lithium concentrations (0.6–1.2 mM). As delineated by York and colleagues, a group of at least four related phosphomonoesterases are inhibited by lithium; these phosphomonoesterases are a group of magnesium-dependent, lithium-sensitive phosphatases that, in mammals, currently includes inositol polyphosphate 1-phosphatase (IPPase), inositol monophosphate phosphatase (IMPase), fructose 1,6-bisphosphatase (FBPase), and bisphosphate nucleotidase (BPNase) (12).
Direct Targets of Lithium
All members of this small group contain a conserved amino acid sequence motif, Asp-Pro-(Ile or Leu)-Asp-(Gly or Ser)-(Thr or Ser), and have a common core tertiary structure that binds metal ions and participates in catalytic functions of the enzyme (12). Of these enzymes, IPPase, IMPase, and FBPase were originally identified as containing this conserved structure (12), whereas BPNase was identified subsequently based upon commonly shared amino acid sequences (13). Newer technology utilizing computer-assisted molecular modeling may allow for more extensive structural characterization of the properties of this binding site, with the potential to discover novel enzymes inhibited by lithium that do not contain this specific motif.
Lithium also inhibits the metabolic enzymes phosphoglucomutase (PGM) (14–17) and glycogen synthase kinase-3 (GSK-3), a serine–threonine kinase that functions as an intermediary in numerous intracellular signaling pathways (18, 19) (Table 1⇑). Significant research effort has focused on IMPase and GSK-3 as possible therapeutically relevant targets of lithium inhibition (Figure 1⇓) based predominantly on the roles these enzymes play in CNS functions (20). Pharmaceutical companies have focused on both of these lithium targets, and it is very likely that a future pharmaceutical inhibitor of either IMPase or GSK-3 may have lithium-mimetic properties in the treatment of bipolar disorder.
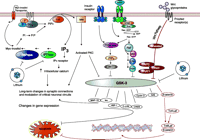
Glycogen synthase kinase-3 (GSK-3) and inositol monophosphatase (IMPase) are direct targets of lithium. This simplified figure highlights relevant interactions among intracellular pathways related to lithium’s action. GSK-3 functions as an intermediary in a number of signaling pathways including neurotrophic signaling pathways, the insulin–phosphatidylinositol 3 kinase (PI3K) pathway and the Wnt pathway—activation of these pathways inhibits GSK-3. The upper left portion of the figure depicts lithium’s actions on the PI signaling pathway. Activation of some G proteins induces phopsholipase C hydrolysis of phosphoinositide-4,5-bisphosphate (PIP2) to diacylglycerol (DAG) and inositol-1,4,5-triphosphate (IP3). DAG activates protein kinase C (PKC). IP3 binds to the IP3 receptor that also functions as a calcium channel in the cell. IP3 is recycled back to PIP2 by IMPase and inositol polyphosphatase phosphatase (IPPase); both of which are inhibited by lithium (21). The inositol depletion hypothesis suggests that lithium exerts its therapeutic actions by depleting free inositol and thus dampening the activation of downstream signaling pathways in neurons (28). Modified and reproduced with permission from (154)
IMPase and IPPase
IMPase and IPPase are enzymes involved in recycling and de novo synthesis of inositol, which is a necessary component of the phosphoinositol (PI) signaling pathway. Many extracellular receptors [such as the serotonin (5-HT)2, α1, and muscarinic (M)1, 3, and 5 receptors] are coupled to the G protein Gq/11, which, through activation of phospholipase C (PLC) mediates the hydrolysis of a cellular membrane phospholipid, phosphoinositide 4,5-bisphosphate (PIP2), to form the second messengers diacylglycerol (DAG) and inositol-1,4,5-triphosphate (IP3) (21, 22). DAG and IP3 subsequently modulate the activity of a multitude of intracellular events (see below).
A number of inositol phosphate phosphatase (IPPase) enzymes are involved in the dephosphorylation (recycling) of IP3 to inositol, a precursor of membrane PIP2 (21). This recycling is necessary to maintain PI-mediated signaling in cell types where inositol is not freely available. The enzyme IMPase catalyzes the final (and rate-limiting) step in the conversion of IP3 into inositol. IPPase removes a phosphate from inositol-1,4-bisphosphate, at the point just prior to where IMPase participates. Both appear to be critical steps in the maintenance of inositol levels and continuation of PI-mediated signaling (23).
Lithium’s direct effect on IMPase (24, 25) and secondarily on IPPase (26, 27) led to the inositol depletion hypothesis of lithium’s action (28, 29) (Figure 1⇑). The inositol depletion hypothesis suggests that lithium exerts its mood stabilizing effect by inhibiting IMPase, decreasing inositol concentrations and thus the amount of PIP2 available for signaling cascades that rely upon this pathway, including but not limited to neurotrophin signaling pathways, receptor tyrosine kinase pathways, and some G protein–mediated signaling (28). It is hypothesized that the brain is particularly sensitive to lithium because of inositol’s relatively poor penetration across the blood-brain barrier (28) or to a reduced ability of specific neuronal populations to transport inositol across their cell membranes (23). Furthermore, based on the noncompetitve inhibition profile of lithium, more active cells and brain regions may be affected to a greater degree (30); however, a recent study suggests that depletion of inositol may not have major effects on PImediated signaling. Specifically, Berry and colleagues found that the reduction of intracellular inositol in the brain sodium–myoinositol transporter (SMIT1) knockout in mice has no effect on PI levels (31).
Although the data are not entirely consistent, lithium does decrease free inositol levels in brain sections and in the brains of rodents treated with lithium (32, 33). Lithium treatment also decreases myoinositol (another form of inositol) in human subjects (34). Thus, it is our contention that, although it was first proposed more than a decade ago (28, 29), the inositol depletion hypothesis remains a viable one for the mechanism of action of lithium. However, no clinically-approved inhibitors of either IPPase or IMPase are available, and therefore it remains difficult to test the inositol depletion hypothesis in patients with bipolar disorder. Past pharmaceutical industry efforts have attempted to develop a brain-penetrating IMPase inhibitor by altering the primary substrate of IMPase—inositol monophosphate (35). Compounds with sufficient inhibition properties were developed, but have thus far failed to advance through clinical trials because they are too highly charged (36), or extremely lipophilic (37), both properties that limit bioavailability in the brain (35). The published crystal structure and modeling studies of IMPase may help to develop novel inhibitors (38, 39). As we discuss below, downstream molecules [notably protein kinase C (PKC)] of IMPase signaling and the PI pathway may also be relevant targets.
GSK-3
GSK-3 is a serine–threonine kinase that is normally highly active in cells, and is deactivated by signals originating from numerous signaling pathways [for example the Wnt pathway, PI-3′ kinase (PI3K) pathway, protein kinase A, protein kinase C, among many others]. It is found in two isoforms, α and β, that have similar, but not always identical, biological functions. Cellular targets of GSK-3 are numerous and often depend on the signaling pathway that is acting upon it (due to cellular localization and regional sequestration). For example, Wnt pathway-mediated inhibition of GSK-3 activates the transcription factor beta;-catenin, whereas in the insulin–PI3K signaling pathway, inhibition of GSK-3 results in activation of the enzyme glycogen synthase. Targets of GSK-3 include, among others, transcription factors [β-catenin, cyclic AMP response element binding protein (CREB), c-Jun], proteins bound to microtubules [Tau, microtubule-associated protein (MAP)-1B, kinesin light chain], cell cycle mediators (cyclin D, human ninein), and regulators of metabolism (glycogen synthase, pyruvate dehydrogenase) (20, 40) (Figure 1⇑).
As a component of many signaling pathways, with multiple cellular targets to choose from, GSK-3 is able to regulate a diverse array of cellular processes such as glycogen synthesis, gene transcription, events related to synaptic plasticity, apoptosis (cell death), and the circadian cycle (41–44). Although many of these functions are likely critically important to both cellular and organism functioning, GSK-3 is currently receiving the most interest as a regulator of apoptosis and cellular resilience (Figure 1⇑). Generally, increased activity of GSK-3 has pro-apoptotic effects, whereas inhibiting GSK-3 attenuates or prevents apoptosis (42, 44).
Evidence suggests an association between mood disorders and impairments of neuroplasticity and cellular resilience—with both in vivo and postmortem studies suggesting neuron and/or glial cell loss or atrophy in circumscribed brain areas (45, 46). Importantly, lithium likely has neuroprotective effects, both clinically and in rodent and cell-based models (45, 47). Lithium may exert these neuroprotective effects at least partly by inhibiting GSK-3 (42, 44).
In 1996, Klein and Melton noted that lithium administration to developing Xenopus embryos had the same effect—duplication of the dorsal axis (48)—as did down-regulation of GSK-3 activity (49). These parallel observations led them to study the direct effects of lithium on GSK-3 (18). Lithium was initially found to inhibit GSK-3 with an enzyme inhibition constant (Ki) of 1–2 mM (serum therapeutic range 0.6 to 1.2 mM) (18, 19); however, evidence showing that lithium inhibits GSK-3 by competing with magnesium (9, 50) suggests that the original studies using higher than physiological levels of magnesium may have underestimated the degree of inhibition.
Early studies suggested that peripheral administration of lithium inhibited brain GSK-3 in the 7-day-old rat brain (51). More recently, studies suggest that this enzyme is significantly inhibited in the rodent brain in the presence of therapeutic serum lithium concentrations during long-term treatment. For example, it was demonstrated that nine days of lithium treatment (at a mean serum concentration of 0.8 mM) of rats increased cytosolic protein levels of β-catenin, a transcription factor regulated directly by GSK-3 (52). This protein level increase was accompanied by a small but significant decrease in β-catenin mRNA levels (reflecting cellular compensation), further suggesting that lithium exerted its actions posttranslationally by inhibiting GSK-3 (52). Confirmatory findings reporting that chronic lithium indeed activates β-catenin-dependent transcription in the mouse brain has been recently published (53). Furthermore, Phiel and colleagues found that three weeks of lithium treatment (at serum levels of 0.8–1.2 mM) decreased the amount of amyloid-β peptide in the brains of AP-Swedish/Tg2576 mice (used in modeling familial Alzheimer Disease), a finding that is likely due to inhibition of GSK-3 (54) given lithium’s effect on accumulation of amyloid- β in cell culture (54–56). These preclinical (i.e., animal or cellular) studies clearly suggest that therapeutic serum concentrations of lithium produce a biologically significant inhibition of GSK-3 in the mammalian brain.
Although GSK-3 was identified in 1996 as the target of lithium responsible for the developmental effects in Xenopus embryos (18), only recently has further evidence been obtained substantially supporting the claim that GSK-3 represents a therapeutic target of lithium. As discussed, GSK-3 represents a strong candidate as a mediator of lithium’s neuroprotective effects most likely because GSK-3 in the brain is significantly inhibited by therapeutic lithium concentrations. Additionally, recent evidence suggests that the behavioral effects of lithium, at least in rodent models, may also be due to inhibition of GSK-3. Three groups have found that administration of GSK-3 inhibitors results in antidepressant-like effects in the forced swim test paradigm following either intracerebral ventricle injections in mice (57), peripheral administration to rats (58), or lithium administration to mice (53). Furthermore, O’Brien and colleagues have recently examined the behavioral effects of knocking out a single copy of the GSK-3β gene, observing in these animals the same antidepressant-like behavior induced by alternate pharmacological inhibition and by lithium administration (i.e., increased mobility in the forced swim test) (53). Further supporting the hypothesis that the effects of antidepressants may be mediated in a GSK-3-dependent manner, Li and colleagues reported that inhibitory phosphorylation (on Ser9) of GSK-3 is acutely increased by increasing the concentrations of 5-HT in the brain through a variety of pharmacological mechanisms (59). Thus, GSK-3 inhibition may represent a therapeutically relevant downstream consequence of antidepressant drugs that initially target serotonin levels.
Amphetamine-induced hyperactivity is the most established rodent model for mania. This behavior is reproducibly attenuated by a number of mood stabilizers including lithium, anticonvulsants, and antipsychotics. Beaulieu et al. recently reported that dopamine-dependent activity increases in mice are mediated in large part via a GSK-3-dependent mechanism (60). They report that both lithium and alternative GSK-3 inhibitors attenuate the hyperactivity in mice lacking the dopamine transporter. They also found that amphetamine administration to wild-type mice results in a decrease in the inhibitory phosphorylation of GSK-3, and that mice heterozygous for GSK-3 have an attenuated response to amphetamine administration. Accordingly, peripheral administration of a GSK-3 inhibitor decreases amphetamine-induced hyperactivity in rats (58). In toto, these data support the possibility that inhibition of GSK-3 may represent lithium’s antimanic as well as its antidepressant target. It will be critical to future understanding of mood disorder etiology to determine which GSK-3 target(s) are responsible for behavior in models of both mania and depression.
In addition to its possible usefulness in the treatment of bipolar disorder (20), inactivation of GSK-3 has been suggested as a potential therapy for a number of diseases, with diabetes and Alzheimer disease receiving the most attention. Diabetes has drawn interest because GSK-3 phosphorylates and deactivates glycogen synthase (61). Alzheimer disease is a target of interest because GSK-3 participates in both the phosphorylation of tau (62, 63) and in the assembly of amyloid-β (54, 55, 64), both of which are thought to be significantly involved in the neurobiology of Alzheimer disease. Specifically, hyperphosphorylation of tau is associated with the formation of neurofibrillary tangles, and accumulation of amyloid-β leads to amyloid plaques. GSK-3 inhibitors may also be useful for the treatment of cardiac ischemic injury (65), baldness and alopecia [the Wnt pathway is involved in hair growth (66)], other neurodegenerative disorders (45, 47) and stroke and other neurotraumatic injuries (47, 67, 68).
FBPase, BPNase, and PGM
Lithium inhibits FBPase, BPNase, and PGM at therapeutic concentrations (10). Fructose-1, 6-bisphosphate (a regulator of gluconeogenesis), removes the 1-phosphate from FBPase to form fructose 6-phosphate. Lithium’s inhibition of FBPase was originally described a number of years ago (14, 69, 70), and more recent studies support these findings (71, 72). Lithium-dependent inhibition of FBPase has not received much attention, however, probably because dysfunction of glyconeogenesis is not a primary theory of bipolar disorder pathophysiology. Inhibitors of FBPase are under development as possible treatments for diabetes (73).
Mammalian BPNase acts on bisphosphorylated nucleotides such as 3′-phosphoadenosine 5′-phosphate (PAP), where it removes the 3′ phosphate to form adenosine 5′-phosphate (AMP) (13, 74, 75); hence, BPNase is also referred to as PAP phosphatase. Sulfotransferases are enzymes that transfer a sulfate group to various biomolecules, using 3′-phosphoadenosine 5′-phosphosulfate (PAPS) as a sulfate donor. PAP is produced following the removal of the sulfate group from PAPS, and acts as an inhibitor of sulfotransferases. Therefore, inhibition of BPNase (and the subsequent buildup of PAP) would be expected to inhibit sulfotransferases. Although studies in mammalian systems are lacking, biochemical reactions potentially modulated by BPNase and/or PAP accumulation include RNA processing metabolism, sodium homeostasis, and sulfation.
The development of nephrogenic diabetes insipidus in patients undergoing lithium therapy might arise from the inhibition of BPNase (13). BPNase, similar to IPPase, hydrolyzes inositol- 1,4-bisphosphate, and lithium prevents BPNase-mediated hydrolysis of both substrates (13, 74, 75). Thus, lithium inhibition of BPNase would be expected to have important effects on inositol recycling, similar to inhibiting IMPase or IPPase. The recently described crystal structure of BPNase should help promote the development of novel inhibitors (76), and a recent review has noted some of the possible roles of BPNase in bipolar disorder (77)
PGM catalyzes the formation of glucose 1-phosphate from glucose 6-phosphate during glycogenolysis (and the reverse during glycogenesis). Lithium was originally identified to inhibit the rabbit and rat PGM enzyme (14–16), and more recently has been found to inhibit human and yeast PGM (17). The role of PGM as a therapeutic target in bipolar disorder treatment has been mostly overlooked due to limited evidence that metabolism of glycogen is involved in this disorder.
Downstream Targets of Lithium
Several signaling pathways exist that are regulated by a number of mood stabilizers (10); those signaling pathways where lithium plays an important role, i.e., the adenylate cyclase (AC), phosphoinositide (PI), arachidonic acid (AA) and neurotrophic-related signaling pathways, are further discussed in detail here.
Cyclic AMP-mediated Signal Transduction
Significant lithium-dependent modulation of cyclic adenosine monophosphate (cAMP)-mediated signaling has been reported. G proteins modulate intracellular cAMP levels by mediating the effect of neurotransmitters (via extracellular receptors) on AC, an integral membrane protein of which there exist numerous subtypes. AC catalyzes the conversion of adenosine triphosphate (ATP) to cAMP. Stimulation of the G proteins Gαs and Gαolf increases AC activity, whereas stimulation of Gαi results in a decrease in AC activity. The physiologic effects of cAMP appear to be mediated primarily by activation of protein kinase A (PKA), an enzyme that phosphorylates and regulates many proteins including ion channels, cytoskeletal elements, transcription factors, and other enzymes. One direct target in the central nervous system (CNS) for the actions of PKA is the transcription factor CREB, which plays a major role in long-term neuroplasticity, and is a downstream target of antidepressants. It is noteworthy that transcription of the CREB gene increases following long-term treatment of rodents with a variety of anti-depressants (22, 78).
One of the genes activated by CREB is brain-derived neurotrophic factor (BDNF), a protein implicated in neuronal survival and synaptic plasticity. There is a growing body of data suggesting that agents that directly modulate the cAMP–PKA–CREB–BDNF signaling cascade may be useful in the treatment of depression (79). In addition to antidepressant effects on cAMP-mediated signaling, mood stabilizers also appear to regulate this pathway. Both lithium and VPA increase BDNF levels in the brains of rats treated chronically with these drugs (80–82). Thus, it is useful to keep in mind that multiple interactions between signaling pathways, e.g., CREB activity and BDNF expression, are regulated by multiple signaling pathways including neurotrophic signaling pathways (as discussed later in this review), and that the cAMP signaling pathway does much more than simply regulate CREB activity.
Lithium appears to have complex effects on cAMP-mediated signaling, with the preponderance of the data demonstrating an elevation of basal AC activity, but also a reduction of receptor-stimulated responses in both preclinical and clinical studies [see (83) for an excellent and thorough review of these data]. Thus, a number of independent research laboratories have found in preclinical models that the ability of the receptor-mediated signal to be propagated via AC is decreased after lithium treatment (22, 83). These extensive cellular findings are consistent with an animal model wherein cholera toxin (a stimulator of the G proteins Gs and Golf) induces hyperactivity when injected into the nucleus accumbens of rats. Cholera toxin-induced hyperactivity was decreased by lithium administration (84), consistent with decreased Gs and/or Golf activity during lithium treatment. But whereas stimulated levels are decreased, there is evidence to suggest an increase in basal cAMP activity (83). These complex, potentially regional specific effects on basal activity and stimulated AC activity may arise from lithium’s effects on G proteins, AC subtypes, and their relative abundance in different brain regions (83).
Postmortem and peripheral cell studies are also consistent with a role of cAMP in mood disorders. Postmortem brain studies of patients who had bipolar disorder reveal increased levels of Gαs and post-receptor stimulated AC activity (85, 86). Generally, the experiments measuring AC activity in unipolar depression find both reduced immediate and long-term effects (87). Thus, although an oversimplification, the majority of the evidence reports increased activity of the AC system in bipolar disorder and a decrease in activity in unipolar depression.
Caution is warranted when attempting to correlate these preclinical and postmortem studies with human disease; however, the available evidence is noteworthy. There are numerous compounds that inhibit AC activity. Particularly, a good deal of specificity has been observed with analogs of the nucleoside adenosine, also called P-site inhibitors (88, 89). Ideally, novel compounds would be isoform-selective in order to avoid peripheral side effects due to the widespread distribution of multiple AC isoforms in different organs in the body. The development of these compounds suggests the eventual possibility of trials with these medications in the treatment of bipolar disorder. Stimulators of AC (e.g., forskolin) may be useful for challenge studies.
PI-Mediated Signaling
Inositol phospholipids play a major role in receptor-mediated signal-transduction pathways, involved in a diverse range of responses such as cell division, secretion, neuronal excitability, and responsiveness. The PI pathway is initiated by the activation of G protein–coupled receptors. M1, M2, M3, α1, and 5-HT2 receptors coupled to Gαq/11 induce PLC hydrolysis of the membrane component PIP2. Hydrolysis of PIP2 by PLC results in the formation of the intracellular second messengers IP3 and DAG, an endogenous activator of PKC. IP3 binds to the IP3 receptor facilitating the release of calcium from intracellular stores, in particular the endoplasmic reticulum (22).
Among other proteins, the Ca2+-receptor protein calmodulin (CaM) stimulates calmodulin-dependent protein kinases (CaMKs) that regulate the activity of diverse proteins, including ion channels, signaling molecules, proteins that regulate apoptosis, scaffolding proteins, and transcription factors (90). As described earlier, IPPase and IMPase (enzymes that are involved in recycling of IP3 back to PIP2) are directly inhibited by lithium (21) (Figure 1⇑). Lithium’s inhibition of these enzymes led to the inositol depletion hypothesis of lithium’s action, which suggests that lithium, via inhibition of IMPase, decreases the availability of myoinositol, and thus the amount of PIP2 available for G protein–mediated signaling events that rely upon this pathway (28).
The inositol depletion hypothesis led to a number of studies, both in cultured cells and in animal models, to determine if the PI pathway may be involved in the pathophysiology or treatment of bipolar disorder (91). Interestingly, a number of studies have suggested the possibility that multiple distinct mood stabilizers may regulate the PI signaling pathway. These include studies of SMIT1, a high affinity myoinositol transport system that has been characterized in various cell types, including those of neural origin (92). The activity and expression of SMIT mRNA in cultured astrocytes is downregulated after chronic treatment with therapeutic concentrations of lithium (92, 93). Decreased expression of SMIT was also observed after VPA or carbamazepine treatment (92, 93). If replicated in vivo, these findings suggest that SMIT may represent a novel target for the development of new drugs.
Another finding implicating PI signaling in the actions of mood stabilizers comes from Williams and colleagues, who used a tissue-culture assay that measures sensory neuron growth-cone stability to conclude that the depletion of neuronal IP3 may be a common mechanism of action of mood stabilizers (94). These investigators demonstrated that lithium, VPA, and carbamazepine all inhibit the collapse of sensory neuron growth cones and increase growth-cone area, effects which were reversed by inositol. The authors then used Dictyostelium, a soil-living organism that relies on IP3 for its development, to identify mutants that confer resistance to the drugs: null mutations of prolyl oligopeptidase confer lithium resistance and elevate intracellular levels of IP3. The authors established a link between lithium and IP3 by showing that prolyl oligopeptidase inhibitors abolished the effects of lithium, carbamazepine, and VPA on growth-cone collapse and area in their tissue-culture assay (94).
PKC and Myristoylated Alanine-Rich C Kinase Substrate (MARCKS)
PKC is a primary target of DAG (Figure 1⇑), and as such, has been an object of intense research in regard to the actions of lithium and other mood stabilizers on the PI pathway. PKC is a ubiquitous enzyme, highly enriched in the brain, where it plays a significant role in regulating both pre- and postsynaptic aspects of neurotransmission (95). Recent studies have suggested that PKC activation may facilitate neurotransmitter release via a variety of mechanisms, including: 1) modulation of several ionic conductances regulating Ca2+ influx; 2) upstream steps regulating release of Ca2+ from intracellular stores; 3) recruitment of neurotransmitter-containing vesicles to at least two distinct vesicle pools; and 4) the Ca2+ sensitivity of the release process itself. PKC is active in many other cellular processes, including stimulating transmembrane glucose transport, secretion, exocytosis, smooth muscle contraction, gene expression, modulation of ion conductance, cell proliferation, and desensitization of extracellular receptors (95).
PKC and PKC signaling appear to be a target of both lithium and VPA (91). Chronic lithium treatment decreases the level of PKC isozymes α and ε (96–98) in cell culture and in treated rodents. The precise mechanisms by which lithium exerts these isozyme-selective actions is unknown, but there is evidence that it is partly due to lithium’s inhibition of IMPase (91, 96). Further evidence supporting the effects of lithium on PKC are data showing that lithium decreases the levels and phosphorylation of a major PKC substrate, myristoylated alanine-rich C kinase substrate (MARCKS), following chronic treatment in rats (99). In cultured cells, this lithium-mediated effect appears to be dependent on low concentrations of inositol in the media, thus implicating lithium’s inhibition of IMPase and/or IPPase as a causative factor (91, 100).
PKC Signaling in Animal Models of Mood Disorders
Current animal models of mania that have been used in the study of mood disorders include kindling, behavioral/amphetamine sensitization, and glucocorticoid administration (43, 101, 102), and PKC activity is implicated in all of these models. Kindling is an animal model for epilepsy that has been proposed to have similarities with pathophysiological aspects of bipolar disorder, in which repeated administration of an electrical stimulus (that is, subthreshold to produce seizures) results in a convulsion and a permanent state of hyperexcitability to the stimulus. These studies on rats have consistently shown hippocampal kindling leads to increased PKC activity and protein concentration (103–108), findings that also were demonstrated to be valid in other brain structures such as the amygdala (109, 110) and neocortex (111, 112).
Studies have also implicated alterations in PKC activity as mediators of long-term alterations in neuronal excitability in the brain following chronic stimulant use. Several independent laboratories have demonstrated that both acute and chronic amphetamine produce an alteration in PKC activity, its relative cytosolto-membrane distribution, as well as the phosphorylation of a major PKC substrate, growth-associated protein (GAP)-43, which has been implicated in long-term alterations of neurotransmitter release (113–117). Furthermore, PKC inhibitors have been shown to block the acute responses (as assessed by both behavioral and in vivo microdialysis studies) to both amphetamine (118) and cocaine as well as cocaine-induced sensitization (119, 120). To further explore the possibility that the PKC signaling may play a role in mood stabilization, a series of studies were undertaken to investigate the behavioral sequelae of PKC inhibition by testing the effects of tamoxifen on three psychostimulant-induced behaviors, representing different validated animal models of mania. Although not a selective agent (better known for its antiestrogenic effects), tamoxifen represents the only CNS-penetrant PKC inhibitor currently available for human use. Tamoxifen significantly reduced acute or chronic amphetamine-induced hyperactivity in a large open field without affecting spontaneous activity levels. However, the same treatment normalized amphetamine-induced increase in visits to the center of an open field (representing risktaking behavior) and reduced hedonic-like amphetamine-induced conditioned place preference (Einat et al., unpublished data). Additionally, recent nonhuman primate studies investigating cognitive deficits similar to those observed in mania have also demonstrated the efficacy of a selective PKC inhibitor (Birnbaum et al., unpublished data).
Thus, although considerable caution needs to be employed when extrapolating from rodent brain and animal behavioral models, the fact that the various animal models of mania are associated with opposite effects on PKC signaling to those observed with chronic lithium or VPA is compelling. In toto, the preclinical data supports further exploration of PKC inhibition as a possible target for new medications. Indeed, CNS-penetrant PKC inhibitors may not only have considerable utility in the treatment of acute mania, but may also exert their effects much more rapidly than existing medications. Such a contention is supported by the findings of a pilot study demonstrating the antimanic effects of tamoxifen (121); large scale clinical trials of PKC inhibitors are clearly warranted.
Neurotrophic Signaling Cascades
Neurotrophins are a family of regulatory factors that mediate the differentiation and survival of neurons, as well as the modulation of synaptic transmission and synaptic plasticity. The neurotrophin family now includes, among others, nerve growth factor (NGF), BDNF, neurotrophin (NT)-3, NT-4, NT-5, and NT-6. BDNF and other neurotrophic factors are necessary for the survival and function of neurons, implying that a sustained reduction of these factors could affect neuronal viability. BDNF also has a number of much more acute effects on synaptic plasticity and neurotransmitter release and facilitates the release of glutamate, γ-aminobutyric acid (GABA), dopamine, and serotonin (122).
BDNF is best known for its long-term neurotrophic and neuroprotective effects, which may be very important for its putative role in the pathophysiology and treatment of mood disorders. Although endogenous neurotrophic factors have traditionally been viewed as increasing cell survival by providing necessary trophic support, it is now clear that their survival-promoting effects are mediated in large part by inhibiting cell death (apoptosis) cascades (122). Increasing evidence suggests that neurotrophic factors inhibit cell death cascades by activating the extracellularregulated kinase (ERK) signaling pathway, the PLC-γ cascade, and the PI3K–Akt pathway. Chronic stress (21 days of foot-shock, an animal model of depression) in rats induced a pronounced and persistent ERK1/2 hyperphosphorylation in dendrites of the higher prefrontal cortical layers of rat brains, whereas phospho-CREB was reduced in several cortical regions including the frontal cortex (123). Because CREB is phosphorylated and activated by phospho-ERK1/2 directly, this phospho-CREB reduction indicates that chronic stress could downregulate CREB phosphorylation indirectly, and subsequently downregulate the transcription of some neurotrophic genes such as bcl-2 and BDNF.
In this context, it is noteworthy that severe stress exacerbates stroke outcome by suppressing bcl-2 expression (124); mice exposed to aggressive social stress expressed approximately 70% less bcl-2 mRNA than unstressed mice following ischemia. Furthermore, stress greatly exacerbated infarct area in control mice, but not in transgenic mice that constitutively express increased neuronal bcl-2. Finally, high corticosterone concentrations were significantly correlated with larger infarcts in wild-type mice but not in transgenic mice overexpressing bcl-2. Thus, enhanced bcl-2 expression appears to be capable of offsetting the potentially deleterious consequences of stress-induced neuronal endangerment, and suggests that pharmacologically-induced upregulation of bcl-2 may have considerable utility in the treatment of a variety of disorders associated with endogenous or acquired impairments of cellular resilience.
Overall, it is clear that the neurotrophic factor–ERK/MAP kinase–bcl-2 signaling cascade plays a critical role in cell survival in the CNS, and that there is a fine balance maintained between the levels and activities of cell survival and cell death factors. Dysregulation of the BDNF–ERK–CREB coordination may be a key mechanism by which prolonged stress induces atrophy of selective subpopulations of vulnerable neurons and/or distal dendrites. Conceivably, the precise kinetics of ERK and CREB activation will ultimately dictate whether the activated kinases participate in a cell survival– or death-promoting pathway.
Neurotrophic Effects of Lithium in Animals
How does the important role of ERK/MAP kinases in mediating long-term neuroplastic events relate to the molecular actions of lithium? Lithium and VPA, at therapeutically relevant concentrations, activate the ERK/MAP kinase cascade in human neuroblastoma SH-SY5Y cells (125) and in critical limbic and limbic-related areas of the rodent brain (80). Neurotrophic factors are now known to promote cell survival by activating MAP kinases to suppress intrinsic, cellular apoptotic machinery, not only by inducing cell survival pathways (122). Thus, a downstream target of the MAP kinase cascade, ribosomal S6 kinase (Rsk) phosphorylates CREB, leading to the induction of bcl-2 gene expression (Figure 1⇑). Consistent with an activation of neurotrophic signaling cascades, chronic treatment of rats with the animal equivalent of therapeutic doses of lithium or VPA produces an increase in the activation of Rsk and CREB, and eventually a doubling of bcl- 2 levels in frontal cortex, effects which are primarily due to a marked increase in the number of bcl-2 immunoreactive cells in layers II and III of the frontal cortex (126–128). Interestingly, the importance of neurons in layers II–IV of the frontal cortex in mood disorders has recently been emphasized, because primate studies indicate that these areas are important for providing connections with other cortical regions, and that they are targets for subcortical input (129).
Further suggestive evidence that lithium and VPA activate the MAP kinase pathway and/or targets of this pathway comes from the data showing that chronic administration of lithium or VPA can increase the expression of BDNF in the rodent brain (80, 81)
Consistent with its effects on neurotrophic signaling cascades, lithium is neuroprotective in animal models of ischemia and Huntington disease can promote neurogenesis in the hippocampus of rats to increase the regeneration of CNS axons (130) and is neuroprotective in many cell culture models (45, 47). Recent evidence suggests that the neuroprotective effect of lithium in cortical neurons requires BDNF expression (131).
Neurotrophic Effects of Lithium in Humans
The body of preclinical data demonstrating neurotrophic and neuroprotective effects of mood stabilizers is striking, yet considerable caution must be exercised in extrapolating these data to the clinical situation with humans. In view of lithium’s robust effects on the levels of the cytoprotective protein bcl-2 in the frontal cortex, Drevets and associates reanalyzed older data demonstrating an approximate 40% reduction in subgenual prefrontal cortex volumes in familial mood disorder subjects (132). Consistent with neurotrophic and neuroprotective effects of lithium, they found that the patients treated with chronic lithium or VPA had subgenual prefrontal cortex volumes that were significantly greater relative to untreated patients, and not significantly different from controls (Wayne Drevets, personal communication). In a more recent study, Drevets and colleagues investigated glial cell densities in mood disordered patients, and although the sample sizes were small, unipolar patients in this study exhibited reduced glial-cell densities, whereas only the bipolar patients who discontinued chronic lithium or VPA exhibited similar reductions (133), suggesting a neuroprotective role associated with the utilization of these agents in patients.
Although the results of the studies noted previously suggest that mood stabilizers may have provided neuroprotective effects during naturalistic use, small sample sizes and the cross-sectional nature of the studies warrant caution. To investigate the potential neurotrophic effects of lithium in humans more definitively, a longitudinal clinical study was recently undertaken using proton magnetic resonance spectroscopy (1H MRS) to measure N-acetylaspartate (NAA, a putative marker of neuronal viability) levels (134). Four weeks of lithium treatment produced a significant increase in NAA levels, effects that were localized almost exclusively to gray matter (135). These findings provide intriguing indirect support for the contention that chronic lithium increases neuronal viability and function in the human brain. Furthermore, a very high correlation (R = 0.97) between lithium-induced NAA increases and regional voxel (i.e., volume pixel, the smallest distinguishable box-shaped part of a three-dimensional image) gray matter content was observed, thereby providing evidence for colocalization with increased expression of bcl-2 in specific regions observed (e.g., gray vs white matter) in the rodent brain cortices. These results suggest that chronic lithium may not only exert robust neuroprotective effects (as has been demonstrated in a vari-ety of preclinical paradigms), but also exerts neurotrophic effects in humans.
A follow-up volumetric magnetic resonance imaging (MRI) study demonstrated that four weeks of lithium treatment also significantly increased total gray matter content in the human brain (136), suggesting an increase in the volume of the neuropil, the moss-like layer comprised of axonal and dendritic fibers that occupies much of the cortex grey matter volume. A finer grained subregional analysis of this brain imaging data is ongoing, but clearly shows that lithium produces a regionally- selective increase in gray matter, with prominent effects observed in the hippocampus and caudate (unpublished observations; G.J. Moore and H.K. Manji). Furthermore, no changes in overall gray matter volume are observed in healthy volunteers treated chronically with lithium, suggesting that lithium is truly producing a reversal of illnessrelated atrophy, rather than non-specific gray matter increases. Recently, cross-sectional studies have corroborated the gray matter findings (137) and NAA findings (138).
Arachidonic Acid (AA) Metabolism
AA functions as an important mediator of second messenger pathways within the brain (139, 140). AA is released from membrane phospholipids via receptor–G protein–initiated activation of phospholipase A2 (PLA2) (141). This action results in release of AA from the cellular membrane, and cyclooxygenase (COX)-mediated production of eicosanoid metabolites such as prostaglandins and thromboxanes. These metabolites mediate numerous subsequent intracellular responses and, due to their lipid permeable nature, transynaptic responses.
AA metabolism as a target of mood stabilizers was originally suggested by studies done by Rapoport, Chang, and colleagues in 1996 and 2001, showing that chronic lithium or VPA treatment of rats results in selective reductions in the turnover rate in the brain phospholipids of AA (142–144). In the case of lithium, the reduction of AA turnover was 80%, and subsequently it was shown that lithium decreased the gene expression and protein levels of an AA-specific PLA2 (i.e., cytosolic PLA2 [cPLA2]) (145, 146) and the protein levels of COX-2 (147). VPA also decreased the turnover of AA by 33% (142), had no apparent effect on cPLA2 protein levels (142), but decreased protein levels of COX-1 and COX-2 (148). Most recently it has been observed that carbamazepine downregulates PLA2 mediated release of arachidonic acid and its subsequent conversion to prostaglandin E2 by cyclooxygenase (149). These findings suggest that effects of mood stabilizers on cell membranes —and specifically AA turnover—might be relevant to the pharmacological action of lithium and VPA (140, 144).
Further general support for the involvement of the AA signaling pathway in bipolar disorder comes from other preclinical studies. Recent studies in rats found that administration of nonselective COX inhibitors indomethacin and piroxicam prevented amphetamine-stimulated locomotor activity (150) and blocked cocaine sensitization (151) ––both rodent models of mania (102). Also, NS-398, a specific COX-2 inhibitor, attenuates restraint stress (a model of depression)–induced oxidative changes (152). The inflammatory hypothesis of bipolar disorder has led to a clinical trial addressing the effect of a specific COX-2 inhibitor as an adjunct treatment in bipolar patients (153).
Conclusions
Bipolar disorder affects approximately 1–3 percent of the world’s population. There has been little progress, however, in developing truly novel drugs for the treatment of bipolar disorder. In fact, most recent additions to the pharmacopeia are brain-penetrant drugs developed for the treatment of epilepsy or schizophrenia (e.g., anticonvulsants such as carbamazepine and antipsychotics such as olanzapine). Thus, there exists a critical need to develop novel approaches for the treatment of bipolar disorder. Although the task of developing novel medications is very difficult, recent insights into lithium’s actions have identified a number of promising and unexpected targets. Moreover, the demonstration of robust neurotrophic and neuroprotetive effects of lithium suggests that one of psychiatry’s oldest treatments may have considerable utility in the treatment of neurodegenerative disorders as well (47, 54).
The pharmaceutical industry has yet to develop brain-penetrant IMPase inhibitors, but GSK-3 inhibitors are rapidly being developed, and we believe these will be invaluable to discern the role of inhibition of GSK-3 in the treatment of bipolar disorder. Tamoxifen, an antiestrogen agent utilized in the therapy of breast cancer, is currently being investigated as an antimanic agent because of its properties as an inhibtor of protein kinase C; initial results from a single-blind study are encouraging, and these are being followed up by large double-blind studies. Mechanisms to enhance neurotrophic pathways are a major focus for the treatment of neurodegenerative disorders, and it is likely that novel medications for this intent may be soon available. We are optimistic that recent novel insights into the mechanisms of action of lithium will ultimately lead to improved medications for the treatment of those who suffer from bipolar disorder.
Acknowledgments
We are grateful for the support of the Intramural Research Program of the National Institute of Mental Heath, the National Association for Research on Schizophrenia and Depression (NARSAD; Young Investigators Awards to JAQ and TDG) and the Stanley Medical Research Institute (HKM).
- © American Society for Pharmacology and Experimental Theraputics 2004
References

Husseini K. Manji, MD, is Chief of the Laboratory of Molecular Pathophysiology at the National Institute of Mental Health, NIH, HHS. He is actively involved in research investigating the molecular and cellular mechanisms of action of mood-stabilizing agents and helped to establish a Mood Disorders Research Unit, which conducts an integrated series of preclinical and clinical studies focusing on signal transduction pathways in unipolar depression and bipolar disorder. Address correspondence to HKM. E-mail manji{at}nih.gov; fax: 301-480-0123.

Todd D. Gould, MD, is an NIMH Seymour S. Kety Memorial Research Fellow at the Laboratory of Molecular Pathophysiology, Mood, and Anxiety Disorders Program of the National Institute of Mental Health, NIH, HHS. His research focuses upon understanding the pharmacology of psychotropic medications, with a particular interest in mood stabilizer targets including glycogen synthase kinase-3.

Jorge A. Quiroz, MD, is Research Fellow at the Laboratory of Molecular Pathophysiology, Mood, and Anxiety Disorders Program of the National Institute of Mental Health, NIH, HHS. His research in translational neurosciences is focused in the investigation of the role of new pharmacological agents that impact cellular resilience and brain neuroplasticity and its clinical application in major depression and bipolar disorder. In addition, Dr. Quiroz is involved in Magnetic Resonance Spectroscopy research (MRS).

