Molecular Targeting in Radiotherapy: Epidermal Growth Factor Receptor
It is estimated that 1.4 million cases of cancer were diagnosed and that 560,000 Americans died from cancer in 2004 (1). Radiotherapy is a widely used local and regional, with respect to the treatment site, mode of therapy in the treatment of cancer. Despite improved methods of radiation delivery, local recurrence accounts for failure of cancer treatment in 20–30% of patients with solid tumors and is associated with another 30–40% of treatment failures. Thus, strategies for improving the efficacy of radiotherapy are urgently being sought. Radiosensitizers and radioprotectors are agents given before or during radiation exposure that lead to potentiation or attenuation of radiation injury, respectively. Unfortunately, only a few of these agents selectively increase the antitumor effects of radiation therapy, thus limiting their clinical application.
Over the past twenty years, advances in molecular biology have increased our understanding of the mechanisms of uncontrolled growth and differentiation, and have identified potential targets in cancer therapy unique to transformed cells. Concomitantly, advances in our understanding of the molecular aspects of radiation biology have revealed that the radiation response is mediated through many of the same molecular processes that are altered in cancer cells. The identification of relevant molecular targets and the better-informed development of pharmacological, biological, and genetic approaches will selectively enhance the radiation killing of tumor cells.
Ionizing radiation (IR) induces multiple cellular and biological effects by either direct interaction with DNA or through the formation of free radical species leading to DNA damage. These effects include induction of cell cycle–specific growth arrest, repair of the damaged DNA, modulation of radical-scavenging proteins, induction of gene mutations, transcriptional and translational changes, malignant transformation, and cell death. Accumulating evidence suggests that irradiation of cells stimulates a series of biochemical and molecular signals. Various components of the IR-inducible signal transduction cascade function as either survival factors participating in cell growth, proliferation and cellular protection, or as cell death-related factors. For example, a balance between antiapoptotic and proapoptotic interactions may direct the fate of the irradiated cell. Furthermore, tumor cells may respond to the cytotoxic agents by launching a compensatory defense system, and resistance to therapy may be acquired at several points along the cell–death pathway. Identification of interactions between IR and a signaling pathway creates an opportunity to target those signaling intermediates to improve the outcome of radiotherapy. Depending on the nature of the interaction, the desired outcome may require preparation of an agonist or an antagonist. The principal goal of combining targeted therapy with IR is to increase the therapeutic index over that achieved with IR alone. This goal may be reached by either increasing the therapy’s antitumor activity and/or decreasing the deleterious effects on normal tissue. The enhancement in antitumor activity may involve increasing tumor radiosensitivity by altering cell cycle progression; abrogation of DNA repair, inducing apoptosis, inhibiting angiogenesis, blocking IR-induced proliferation; and/or potentiating effective immunologically mediated host responses. Decreasing normal tissue toxicity, particularly late effects, such as radiation-induced pneumonitis or neurotoxicity, may involve increasing normal tissue radioresistance, attenuating detrimental host responses such as fibrosis and bolstering beneficial host responses such as revascularization.
Epidermal growth factor (EGF) and the EGF receptor (EGFR) are involved in normal development and differentiation of epithelial cells as well as in carcinogenesis (2). The most common mutant variant of EGFR in malignancies is the truncated EGFRvIII, resulting from variable intragenic deletions in the 5′ region of wild-type (wt) EGFR involving exons 2–7, with subsequent deletion of 801 bases––a region that encodes amino acids 6–273 of the wt EGFR extracellular domain (3) and creates a new inframe glycine residue at the junction site. Structurally, EGFRvIII lacks most of the extracellular subdomain I and a considerable portion of subdomain II. Subdomain III is the major ligand-binding domain, whereas subdomains II and IV are similar cysteinerich domains involved in intramolecular interactions. Despite the physical presence of subdomain III, EGFRvIII is incapable of binding EGF or tumor growth factor–α (TGFα); however, EGFRvIII is constitutively activated and phosphorylated. In malignant tumor cells, overexpression of wt EGFR has been associated with uncontrolled proliferation, anchorage-independent growth, and tumorigenesis. With respect to the radiation response, EGFR overexpression has been associated with increased radioresistance. EGFR is a member of the ErbB family of receptor tyrosine kinases (RTK). Signaling intermediates include Ras, Raf, mitogen- activated protein kinase (MAPK), phospholipase Cγ (PLCγ), and protein kinase C (PKC) (Figure 1⇓). Moreover, there is substantial evidence that there is crosstalk between EGFR and different receptors. EGFR activation occurs through at least three mechanisms: ligand-induced receptor activation, ionizing radiation, and overexpression (Figure 2⇓). “Overexpression” can take the form of either amplification of the EGFR gene or expression constitutively active mutant forms of the RTK activity. Irrespective of the mechanism of receptor activation, the results are the same, with enhanced proliferation and increased radioresistance (4).
The recent demonstration of EGFR-(also termed ErbB1)-activation by IR makes this receptor and other members of the ErbB family important targets for novel radiosensitizing therapeutic interventions. The IR-induced activation of EGFR results in cytoprotective signaling dominantly involving MAPK and phosphoinositide-3-kinase (PI3K)—cytoplasmic protein kinases that mediate proliferative and biosynthetic responses (Figure 1⇓), respectively. Based on the obligatory link between radiation-induced activation of EGFR and MAPK–PI3K, Schmidt-Ullrich and colleagues have suggested that this response represents the underlying mechanism of radiation-induced accelerated proliferation (5). This response occurs in carcinoma cells whose growth is regulated, in an autocrine manner, by EGFR and other ErbB-family members. Thus, disruption of the tyrosine kinase activity of EGFR should result in radiosensitization of a variety of neoplastic cells, including squamous cell carcinoma (SCC), mammary (MCC), small cell lung (SCLC), and malignant glioma (MGC) cells—all expressing significant levels of EGFR and other ErbB molecules. Schmidt-Ullrich and colleagues have demonstrated that the inhibition of EGFR function by tyrphostins, a group of compounds that inhibit RTK activity, or through overexpression of dominant-negative (DN) EGFR variant (EGFR-CD533) inhibits the radiation-induced proliferation response after single or repeated radiation exposures in the 1–5 Gy dose range (6). In addition, disruption of EGFR function in SCC, MCC, and MGC directly enhances radiosensitivity. Therefore, both the inhibition of the radiation-induced proliferation and the direct radiosensitization contribute increased radiosensitivity in vivo after repeated radiation exposures (7).
The disruption of EGFR function can be accomplished by at least three different approaches (Figure 3⇓). First, monoclonal antibodies (mAbs) directed against the receptor can inhibit its function. Among those studied, mAb C225 inhibits the binding of EGF or TGFα and interferes with the physiological processing of the receptor. Additionally, Mab C225 inhibits tumor cell proliferation, promotes chemo- and radiosensitization, and inhibits tumor growth in vivo by disrupting EGFR-TGFα–dependent angiogenesis in tumor cells (8–11). EGFR-specific mAbs are also being used to direct the delivery of radionuclides, toxins, and immunomodulatory molecules.
The second major approach to disrupting EGFR function involves chemical inhibition of the ErbB1 RTK domain that is critical for transactivation of receptors after growth factor–induced homo- or heterodimerization. These compounds are competitive substrates for the tyrosine kinase in the cytoplasmic, C-terminal domain of the receptor. Compounds from Parke-Davis (CI1033) and AstraZeneca (ZD1839) act to sensitize tumor cells to cytotoxic drugs and to radiation (12–14). Although these experiments have generated impressive results, studies on the mechanisms underlying these effects have been limited until recently. The most attractive development in tyrphostin research is the new generation of inhibitors that possess broad-spectrum activity towards the ErbB family (e.g., CI1033) and irreversibly bind to the kinase domain in vitro; however, the effects of these compounds may be less sustained in cells and thus, less effective in vivo because of ErbB receptor turnover. This possibility highlights the potential benefit of developing a genetic approach that would result in the continuing renewal of the inhibitory factor(s).
Genetic transfer approaches have recently become available for therapeutic use. Inhibition of EGFR expression by antisense oligonucleotides is an effective approach to inhibiting tumor growth in vitro. Another gene-based strategy is based on the overexpression of EGFR-CD533. This DN EGFR variant lacks the C-terminal 533 amino acids and forms nonfunctioning heterodimeric complexes with wt EGFR receptors (15). Schmidt-Ullrich and colleagues have studied the potential therapeutic application of EGFR-CD533 in human tumor xenograft models (16). They have shown that overexpression of EGFR-CD533 in mammary carcinoma cells abrogates the EGFR-mediated cytoprotective response induced by IR and enhances radiosensitivity (17). They subsequently incorporated EGFR-CD533 into a replication-incompetent adenovirus (Ad) and showed that Ad-EGFR-CD533 delays growth of U-373 MG glioma xenograft tumors and overcomes the enhanced tumorigenic effects of EGFRvIII expression (18).
Another significant issue in the use of molecular strategies targeted at the EGFR is the ability to deliver the therapeutic agents to the tumor cells with adequate doses and appropriate timing. Small molecule inhibitors, such as the tyrphostins, present concerns with respect to systemic toxicities, whereas macromolecular and particulate agents––such as the mAbs and adenoviral vectors, respectively––require, specific targeted delivery techniques. A useful approach in this regard is the technique of direct intraparenchymal infusion, a technique that has been used in animal models of head and neck cancer, and has been developed and used more extensively in central nervous system applications (19), where the therapeutic agent accesses the extracellular interstitial space. Although viral particles present additional challenges for adequate distribution to wide areas containing tumor cells, modifications of the infusion techniques, and potential use of carefully designed replication-competent viral vectors have substantial potential for overcoming such difficulties.
As our understanding of molecular radiation biology improves, the number of potential molecular targets will undoubtedly expand as will the number of permutations possible in combining targeted therapy with either IR or chemotherapy. Further investigation of the mechanisms of interactions between IR and signaling networks are needed to exploit the potential of such combinations therapeutically. This knowledge will also be critical to the development of intermediate markers of tumor response that can be applied to cancer therapeutics and predict for clinical response. Ultimately, identification of patient-specific molecular targets may allow for a custom-tailored approach to cancer therapy.
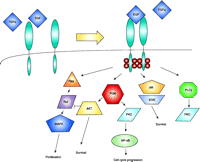
The Epidermal Growth Factor Receptor (EGFR) signaling network. The EGFR is activated upon ligand binding [either EGF or Tumor Growth Factor–α (TGFα)], which promotes homo- or heterodimerization, autophosphorylation (small spheres located on the cytoplasmic domains of receptors), and internalization. Signaling intermediaries include Ras–Raf–MAPK (for proliferation response), PI3K–AKT (for antiapoptotic response), and PKC (for cell cycle progression). PI3K, phosphoinositide 3-kinase; Jak, Janus kinase; STAT, signal transducer and transactivator; PKC, protein kinase C; PLC, phospholipase C; MAPK, mitogen-activated protein kinase; NF-κB, nuclear factor–kappa B.
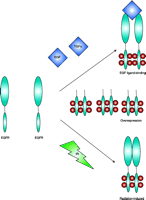
EGFR activation. EGFR can be activated by a number of mechanisms. EGF ligand binding to the EGFR leads to dimerization, internalization, and autophosphorylation. Overexpression of EGFR activity can occur through either gene amplification or constitutively activated mutant forms of EGFR. Finally, ionizing radiation (IR) can activate the EGFR independent of ligand.
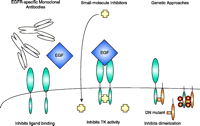
Approaches to disrupting EGFR signaling. At least three approaches are currently studied to target the EGFR. EGFR-specific monoclonal antibodies compete with ligand and behave as receptor antagonists. EGF-specific antibodies are also being used to direct the delivery of radionuclides, toxins, and immunomodulatory molecules. Small-molecule inhibitors interact with the intracellular domain of EGFR and block the TK (tyrosine kinase) activity. Genetic approaches include antisense oligonucleotides to inhibit EGFR expression and overexpression of dominant negative (DN) mutants that form heterodimers with wt EGFR or constitutively activated EGFR.
- © American Society for Pharmacology and Experimental Theraputics 2005
References

William C. Broaddus, MD, PhD, is Associate Professor of Neurosurgery at the Virginia Commonwealth University, Richmond, VA. His research interests include molecular biology and therapy of brain tumors, stereotactic neurosurgery, and the delivery of therapeutics into the central nervous system by direct infusion techniques. E-mail wcbroadd{at}vcu.edu; fax 804-828-4493.

Theodore D. Chung, MD, PhD, is Associate Professor of Radiation Oncology at the Virginia Commonwealth University, Richmond, VA. Dr. Chung’s research focuses on prostate cancer biology, the interaction of radiation with signaling pathways and the clinical application of gene therapy. E-mail tchung{at}hsc.vcu.edu; fax 804-828-6042.

