The NAADP Receptor: New Receptors or New Regulation?
Nicotinic acid adenine dinucleotide phosphate (NAADP) is structurally related to the well-known coenzyme nicotinamide adenine dinucleotide phosphate (NADP), the only difference being the replacement of the nicotinamide moiety by nicotinic acid. In contrast to NADP, NAADP is a potent activator of Ca2+ release from intracellular stores, and in some cases also Ca2+ influx. Such effects have been reported in a wide range of cells and preparations across the phylogenic spectrum. We discuss below on the basis of current work whether NAADP acts on a novel Ca2+-release mechanism or regulates known Ca2+-release channels by novel mechanisms.
The chemical synthesis of NAADP was first described by Bernofsky, who prepared it through an enzymic base-exchange reaction by incubating NADP and nicotinic acid with calf spleen NAD glycohydrolase (1). It was not until 1987, however, that the potent Ca2+-releasing activities of NAADP were reported, as an unidentified contaminant of commercially available NADP (2), which released Ca2+ from vesicular stores in sea urchin egg homogenates. The amount of this contaminant was increased by treating NADP with alkali. Subsequent analysis by HPLC and high resolution mass spectroscopy identified the Ca2+-mobilizing compound as NAADP (3).
The initial studies in sea urchin egg homogenates of alkali-activated NADP-evoked Ca2+ release (2) and, once identified, NAADP-induced Ca2+ release (3, 4), indicated that the release mechanism did not involve the inositol-1,4,5-trisphosphate (InsP3) receptor (IP3R). Indeed, NAADP-, cyclic adenosine diphosphate ribose (cADPR)-, and InsP3-sensitive Ca2+-release mechanisms exhibit specific desensitization, in that, regardless of the order of addition, each agent added at its maximal concentration renders homogenates or microsomes insensitive to further Ca2+ release by the same agent, but fully sensitive to the others (3). These results suggested that there were at least three separate Ca2+ release mechanisms present in sea urchin homogenates.
Pharmacological experiments using selective inhibitors of IP3Rs such as heparin, and those of ryanodine receptors (RyRs), for example ryanodine, ruthenium red and procaine (as well as inhibitory structural analogs of cADPR), indicated that whereas cADPR most likely activates RyRs, NAADP activates a Ca2+-release mechanism that was most likely distinct from IP3Rs and RyRs. Similar results were observed in intact sea urchin eggs injected with IP3Rs or RyR inhibitors in response to photolysis of caged NAADP (5). Although the pharmacology of NAADP-induced Ca2+ release has not been extensively studied, NAADP-sensitive Ca2+-release channels are blocked, noncompetitively, by concentrations of dihydropyridines, gallopamil (also termed methoxyverapamil or D600), or diltiazem that, although being somewhat higher than those required to block voltage-gated Ca2+ channels (Table 1⇓), nevertheless do not affect Ca2+ release via either IP3Rs or RyRs (6).
A hallmark of both IP3R- and RyR-mediated Ca2+ release is that both Ca2+ release channels are modulated in such a way by Ca2+ itself that they are said to mediate Ca2+-induced Ca2+ release (CICR), a process thought to underlie many of the complex spatio-temporal complexities of Ca2+ signaling patterns widely observed in all cell types (7). In contrast, in sea urchin egg homogenates, the NAADP-sensitive Ca2+-release mechanism is insensitive to Ca2+, and resistant to block by millimolar concentrations of Mg2+, which block CICR (6, 8). Thus, in intact sea urchin eggs, NAADP induces localized Ca2+ release, which can lead to regenerative Ca2+ waves, but only if IP3Rs and RyRs are subsequently recruited (5).
Early studies of Percoll-gradient fractionation of alkali-activated NADP-induced Ca2+ release showed that this mechanism regulated release from a nonmitochondrial store devoid of cytochrome c activity, which was separate from those activated by InsP3 or cADPR (generated from NAD+) (2). IP3Rs and RyRs are well-characterized as mainly residing in (sarco-) endoplasmic reticulum (ER) Ca2+ stores, the site of Ca2+ sequestration by thapsigargin-sensitive sarco-endoplasmic reticulum Ca2+ ATPases (SERCA pumps). Whereas thapsigargin, as expected, depletes InsP3-sensitive and cADPR-sensitive Ca2+ stores in both sea urchin egg homogenates (6) and intact eggs (9), NAADP-sensitive stores remain largely unaffected by this treatment. Further evidence for NAADP-sensitive stores being discrete from the ER comes from elegant experiments using stratified sea urchin eggs. Centrifugation of eggs results in the formation of elongated structures in which organelles are stratified in distinct regions. Fluorescence-tagged thapsigargin labels structures at the nuclear pole, which is also the site of both InsP3- and cADPR-induced Ca2+ release. In contrast, NAADP-sensitivity, functionally visualized by Ca2+ imaging, is located at the opposite pole enriched in mitochondria (10) and acidic compartments (11). NAADP-sensitive stores are unlikely to be mitochondria because they express a paucity of mitochondrial markers, thus, acidic organelles emerged as plausible candidates. More-detailed Percoll fractionations of sea urchin egg homogenate allowed the purification of NAADP-sensitive Ca2+ stores with minimal InsP3 or cADPR sensitivity. These stores colocalized with egg reserve granules—functionally equivalent to secretory lysosomes—which are also involved in plasma membrane repair following mechanical wounding (12, 13), providing one explanation for the presence of NAADP-sensitive Ca2+ influx pathways in the plasma membrane (14–16). These stores are enriched in [32P]-NAADP binding sites and lysosomal markers, and sequester Ca2+ which may be discharged by NAADP (11). The vacuolar proton pump inhibitor bafilomycin and various protonophores abrogate this Ca2+ sequestration, and the lysomotropic agent glycyl-l-phenylalanine-β-naphthylamide (GPN) abolishes NAADP-induced Ca2+ release. In intact eggs, as well as in egg homogenates, GPN and bafilomycin selectively abolish NAADP- but not InsP3- or cADPRevoked Ca2+ release (Table 1⇓). Taken together, these data are consistent with a mechanism whereby NAADP-sensitive release is located in thapsigargin-insensitive acidic Ca2+ stores (11).
The nature of the NAADP receptor in sea urchin egg homogenates has been probed by characterizing [32P]-NAADP–binding proteins. Binding is essentially irreversible in this preparation, and, together with the high binding capacity, make it a system of choice for the purification and molecular characterization of these proteins. [32P]-NAADP binds to a single population of high affinity binding sites (Kd ~ 0.3 nM, Bmax ~ 125 fmol/mg protein) on sea urchin egg homogenate membranes (17, 18). Detergent-solubilized [32P]- NAADP–binding proteins display similar binding characteristics to those found in native membranes, indicating that they are intrinsic membrane proteins, and gel-filtration molecular size analysis indicates that they are substantially smaller than either native IP3Rs or RyRs (19).
NAADP-induced Ca2+ release is also widespread in mammalian systems. The exocrine pancreatic acinar cell was the first intact mammalian cell type in which NAADP-induced Ca2+ release was reported (20), but studies on mammalian exocrine cells, in general, have also been crucial for developing our current view of intracellular Ca2+ signaling events. Experiments on the perfused cat submandibular gland provided the earliest evidence for agonist-elicited release of Ca2+ from non-mitochondrial intracellular stores, also demonstrating cellular extrusion of Ca2+ by active transport and, after a delay, activation of Ca2+ entry (21). Studies on permeabilized pancreatic acinar cells demonstrated for the first time that InsP3 releases Ca2+ from a non-mitochondrial store (22).
Combining patch-clamp whole-cell recording of Ca2+-activated Cl− current with fluorescence measurement of the cytosolic Ca2+ concentration (23, 24) has made it possible to probe directly the action of various intracellular messengers in pancreatic acinar cells (23–26). In this way, InsP3 (25, 27), cADPR (26), and NAADP (20, 24, 28) were shown to elicit repetitive local cytosolic Ca2+ spikes in the apical granular pole. The local apical Ca2+ spiking depends on concerted activity of RyRs and IP3Rs (28, 29).
The two physiological activators of pancreatic acinar secretion, acetylcholine (ACh) and cholecystokinin (CCK), initiate Ca2+ spiking via different messenger pathways. Inactivation of NAADP-evoked Ca2+ release in mammalian cells occurs at high concentrations of NAADP (20). This is in contrast to the situation in sea urchin eggs, where concentrations that are below the threshold for inducing Ca2+ release fully desensitize the release mechanism (14). Inactivation of NAADP receptors by intracellular application of NAADP at a high concentration (μM) blocks Ca2+ spiking elicited by physiological CCK concentrations (1–10 pM) (20, 28), but has no effect on Ca2+ spiking responses elicited by ACh (28) or bombesin (30). Physiological Ca2+ signaling elicited by CCK, therefore, has a unique requirement for functional intracellular NAADP receptors.
How and from which intracellular store does NAADP elicit Ca2+ release in the acinar cells? This is difficult to answer on the basis of studies in intact acinar cells, because the apical granular pole, where the NAADP-induced Ca2+ spiking occurs, contains two major Ca2+ pools located in the endoplasmic reticulum (ER) and the zymogen granules (ZGs) (31). The ER is generally considered to be the main Ca2+ store from which cytosolic Ca2+ signals are generated (32), therefore, it would seem desirable to test the action of NAADP on a pure ER preparation. The nucleus may, in practice, constitute such a preparation. The outer nuclear membrane is continuous with the ER membrane and the lumen of the nuclear envelope is continuous with the ER lumen (33). Studies on isolated liver nuclei have demonstrated active, ATP-dependent and thapsigargin-sensitive Ca2+ uptake into the nuclear envelope, and InsP3 and cADPR liberate Ca2+ from this store (34). More recent studies on isolated nuclei from pancreatic acinar cells, which also contain several layers of surrounding ER, demonstrate that the thapsigarginsensitive Ca2+ store can release Ca2+ in response to stimulation, not only with InsP3 or cADPR, but also with NAADP (35). Data obtained from this preparation show that the Ca2+ liberation elicited by NAADP (and also by cADPR) can be blocked by ryanodine or ruthenium red, but not by caffeine. Alternately, the Ca2+ release evoked by InsP3 is blocked by caffeine (acting as an IP3R antagonist), but not by ryanodine or ruthenium red (35). Therefore, the simplest hypothesis to account for these data is that NAADP, like cADPR, causes Ca2+ release from the ER by opening RyRs. NAADP and cADPR act on separate receptor sites, because inactivation of NAADP receptors by a high NAADP concentration (μM) does not prevent cADPR-elicited Ca2+ release (35).
In pancreatic acinar cells, CCK and NAADP can also release Ca2+ from an acidic, bafilomycin-sensitive, but thapsigargin-insensitive, Ca2+ store located in the apical granular pole (36) that new, as yet unpublished results suggest, does not have characteristics of the ER. Ca2+ can be released from this acidic granular store by either InsP3, cADPR, or NAADP. As is the case for the ER, the actions of NAADP and cADPR on the acidic store can be abolished by RyR inhibitors, whereas the action of InsP3 can only be blocked by IP3R antagonists. It has been proposed that the acid pool from which NAADP releases Ca2+ could be in lysosome-related organelles, including ZGs (36). The ZGs have an extraordinarily high total Ca2+ concentration (>10 mM) (37) and isolated single ZGs can release Ca2+ in response to both InsP3 or cADPR (37). So far, the action of NAADP has not been tested on ZGs. Because cytosolic Ca2+ signals generated by stimulation with InsP3, cADPR, or NAADP are remarkably similar (24), it seems possible, even likely, that NAADP could also liberate Ca2+ from isolated ZGs.
Apical Ca2+ signals are of strategic importance for the control of pancreatic enzyme and fluid secretion (38, 39). It appears that the reason for the particular sensitivity of the apical region of pancreatic acinar cells to Ca2+-releasing stimuli is the close proximity, in this part of the cell, of two separate Ca2+ stores, both having a full complement of Ca2+-releasing channels. Given the fact that CICR only initiates in the apical pole and depends on the dual function of both RyRs and IP3Rs (29), it does not seem unreasonable to propose that the apical Ca2+ spikes arise from CICR interactions between these receptors on both types of Ca2+ stores.
Although sea urchin eggs and pancreatic acinar cells are the most intensely studied systems in which the actions and role of NAADP in Ca2+ signaling have been investigated, there is a growing list of other cell types in which NAADP participates as an important Ca2+-releasing messenger. Rat brain microsomes were the first mammalian preparation in which NAADP-induced Ca2+ release was demonstrated (40). In this preparation, the NAADP-regulated Ca2+ release mechanism, as in sea urchin eggs, was independent of extravesicular Ca2+ concentrations, was unaffected by IP3R and RyR inhibitors, but was inhibited by micromolar concentrations of dihyropyridines or diltiazem. These findings were mirrored in studies on cholinergic motoneurons from Aplysia, where NAADP-evoked Ca2+ release was not blocked by heparin or by ryanodine either alone or together (41). Similarly in frog neuromuscular preparations, NAADP-induced increases in transmitter release from prejunctional neurons were not affected by either ryanodine or thapsigargin (42, 43). In rat cortical neurons, NAADP-evoked increases in Ca2+ were thapsigargin- insensitive but again blocked by bafilomycin (44); however, NAADP-induced Ca2+ transients were reduced but not abolished by ryanodine or heparin treatments. These data are consistent with the suggestion that NAADP activates a distinct Ca2+ release mechanism on acidic compartments, that can, in turn, be amplified by recruiting IP3Rs and RyRs, presumably by CICR.
Pancreatic beta cells also express an NAADP-sensitive Ca2+-release mechanism. In human pancreatic beta cells, NAADP mobilizes Ca2+ from a pool that is insensitive to either ryanodine or the IP3R antagonist, xestospongin C, and might mediate the autocrine inhibitory effects of insulin on further release of this hormone (45). In this study, and in a report of the pancreatic beta MIN6 cell line (46), nanomolar concentrations of NAADP induced Ca2+ release, whereas micromolar concentrations inactivated the release mechanism. Because micromolar concentrations of NAADP blocked glucose-induced Ca2+ spiking and because glucose administration increased NAADP levels, a messenger role was proposed for NAADP in stimulus-secretion coupling in pancreatic beta cells (46, 47). These findings were extended in MIN6 cells to show that NAADP mobilizes Ca2+ through a thapsigargin- and ryanodine-insensitive mechanism in secretory granules (SGs) rather than in the ER (48). These results were complemented by a study in intact MIN6 cells, in which NAADP-evoked Ca2+ release was blocked by bafilomycin but not thapsigargin (36).
Smooth muscle is another system in which NAADP-evoked Ca2+ release has been observed. In intact pulmonary arterial myocytes, NAADP mobilizes Ca2+ from a bafilomycin-sensitive store. Whereas xestospongin C had little effect on NAADP-induced Ca2+ release, both ryanodine or thapsigargin reduced but did not abolish NAADP’s effects (49, 50). Thus, as in other systems, local NAADP-induced Ca2+ release from acidic stores might be amplified by CICR through the recruitment of RYRs on the ER located proximal to NAADP-activated receptors (50). In rat aortic microsomes, NAADP released Ca2+ by a mechanism insensitive to IP3R or RyR blockers but which was inhibited by dihydropyridines (51). In striated muscle, NAADP released Ca2+ from rat heart microsomes, which was unaffected by ryanodine and divalent cations, but blocked by dihydropyridines (52); however, it should be noted that direct interactions of NAADP with purified RyRs from both skeletal (53) and cardiac muscle (54) have been reported by some but not others (55).
Interactions between Ca2+-release mechanisms are important in T-cell activation. NAADP evokes Ca2+ release with a concentration- dependence similar to that evinced in both pancreatic acinar and beta cells: nanomolar concentrations activated, but micromolar concentrations inhibited Ca2+ release (56). Through the use of high, inactivating NAADP concentrations, T cell receptor–mediated activation was inhibited as was, perhaps more puzzlingly, Ca2+ release by InsP3 or cADPR. A follow-up study showed that knock-down studies of RyRs (by utilizing antisense mRNA technology) in Jurkat cells— and experiments that inhibited RyRs with ruthenium red—reduced but did not totally abolish NAADP-induced Ca2+ release (16). Based on these studies, it is difficult to distinguish between a direct effect of NAADP on RyRs or an indirect effect involving amplification of NAADP-induced Ca2+ release by RyRs. There is a single report that NAADP may also operate as a Ca2+-mobilizing messenger in plants. In both red beet and cauliflower, it appears that NAADP releases Ca2+ from the ER rather than vacuolar membranes (57) through a mechanism insensitive to either heparin or 8-NH2-cADPR or dihydropyridines, but blocked by ruthenium red, suggesting that it activates a novel Ca2+-release mechanism.
There are now numerous studies showing that NAADP is an extremely potent Ca2+-mobilizing messenger as well as a regulator of Ca2+ influx in a wide range of cell types from a variety of organisms including mammals and humans (Table 1⇓). NAADP was initially implicated as a Ca2+-mobilizing messenger for different agonists in several cell types through the use of protocols that inactivated NAADP-mediated Ca2+ release; these findings have now been confirmed by direct NAADP measurements (14, 46, 50). The precise biosynthetic pathway for NAADP is unclear, although CD38 can catalyze synthesis by a base-exchange mechanism in vitro (58). Another major uncertainty is the nature and cellular location of NAADP-sensitive Ca2+-release channels. Although in many studies, NAADP-evoked Ca2+ release may be insensitive to IP3R and RyR blockers but inhibited by agents such as dihydropyridines, more often than not RyR inhibitors reduce NAADP-mediated Ca2+ release. However, because different Ca2+-release channels often interact to shape Ca2+ signaling patterns (29, 59), it is not trivial to distinguish between a direct action of NAADP at RyRs or an action at a distinct and novel Ca2+-release channel followed by amplification by RyR-mediated CICR (Figure 1⇓). If a direct action is the case, then there is a certain degree of novelty and selectivity as to the mechanism of regulation. If the latter scenario proves correct, then it is likely that we have been confounded by variations in the tightness of coupling between the separate mechanisms in different cells. Another possibility is that NAADP-binding proteins, although likely to be intrinsic membrane proteins (19), are promiscuous and are not intrinsic Ca2+-release channels, but regulators of known Ca2+ channels such as RyRs, and dihydropyridine-sensitive Ca2+ channels, among others. This hypothesis may also explain the apparent heterogeneity of Ca2+ stores that NAADP apparently discharges, with NAADP modulating the Ca2+ channels found in these different subcellular locations. These scenarios will only be unequivocally resolved by the purification and characterization of NAADP-binding proteins.
Pharmacology of the Inhibition of NAADP-Induced Ca2+ Release
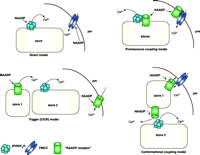
Possible mechanisms for NAADP-induced calcium release.Direct model: NAADP binds directly to the RyR on Ca2+ stores (ER or acidic compartments) increasing the probability of the channel being open (53, 54). NAADP may also bind and directly regulate plasma membrane (pm) Ca2+ channels (PMCC). Trigger CICR model: This model assumes that there is a discrete NAADP receptor/Ca2+-release channel that is located in a discrete Ca2+-storage organelle (probably an acidic store) (store 1). Localized Ca2+ release from this pool then recruits IP3Rs and RyRs located in a second store [the ER (store 2)] by CICR and globalizes the Ca2+ signals (5, 20). Promiscuous coupling model: In this model, the NAADP-binding protein is not a Ca2+ channel per se, but rather is an integral membrane protein that can associate and regulate Ca2+-release channels such as RyRs (35), in the ER or acidic stores (including secretory granules), or calcium influx channels in the plasma membrane. Conformational coupling model: This model assumes that NAADP interacts directly with a distinct NAADP-regulated Ca2+-release channel in a discrete store. This channel can release Ca2+ in response to NAADP, but the channel may also directly interact with other Ca2+-release channels (IP3Rs or RyRs) or with plasma membrane Ca2+-influx channels. This model provides one explanation for how desensitizing concentrations of NAADP apparently render IP3Rs or RyRs insensitive to their respective agonists (56) or desensitize Ca2+ influx (14), even in the apparent absence of Ca2+ release. Permutations of any of the four models above are also possible. Another possibility is that distinct subtypes of NAADP receptors exist with different localizations, pharmacology, and functions.
- © American Society for Pharmacology and Experimental Theraputics 2005
References

Ole H. Petersen, FRS, is Medical Research Council Professor in Physiology, at The University of Liverpool, UK. E-mail o.h.petersen{at}liv.ac.uk; fax +44 151 794 5323.

Antony Galione, PhD, is a Professor of Pharmacology and a Wellcome Senior Fellow in Basic Biomedical Science at The University of Oxford, UK. E-mail antony.galione{at}pharm.ox.ac.uk; fax +44 1865 271 853.

