REGULATION OF DRUG TRANSPORTERS: DURING INFECTION AND INFLAMMATION
Abstract
Inflammation-induced changes in the pharmacokinetics and dynamics of numerous drugs have been reported. Altered drug disposition during inflammatory disease has traditionally been ascribed primarily to changes in drug metabolism and protein binding. Emerging evidence within the last decade, however, has demonstrated that the inflammatory response affects the expression of several important drug transporters and these changes significantly impact the disposition and activity of drug substrates.

Introduction
Pathophysiological changes in patients and animal models of infectious or inflammatory conditions are associated with immediate and often dramatic alterations in the production of numerous liver-derived proteins. Collectively known as the acute phase response (APR), such inflammation-associated changes result in pronounced increases in the plasma concentrations and toxicity of various drugs (1–5). A classic clinical example is the severe toxicity associated with the administration of theophylline to patients with influenza; decreased expression of the cytochromes P450 (P450) reduces the clearance of theophylline, subsequently resulting in toxic accumulation of drug. Interest was further stimulated by clinical reports that inflammatory diseases such as Crohn’s Disease, celiac disease, and rheumatoid arthritis were associated with large increases in plasma drug concentrations after oral doses of propranolol (1) and oxprenolol (2). These findings are not restricted to chronic inflammatory conditions, as viral infections also affect drug disposition. Mild viral infections in otherwise healthy subjects result in 75–100 percent increases in the area under the plasma concentration time curve (AUC) of oxprenolol (2). Early explanations attributed this to an inflammation-mediated reduction in drug clearance. Over the past thirty years, many studies have examined the underlying mechanisms responsible for this phenomenon. Much of the work to date ascribes these changes to alterations in drug metabolizing enzymes and drug-plasma binding proteins (1–5).
Many drugs are actively transported across epithelial membranes during the process of absorption, distribution, and clearance; thus, changes in the expression of transport proteins are also likely to contribute to altered drug disposition in inflammation. Drug transporters are a class of membrane proteins involved in transport of numerous drugs, metabolites, and xenobiotics. Because these membrane transporters are highly expressed in epithelia of the intestine, liver, kidney, placenta, and blood-brain barrier, they serve a major role in defining the pharmacokinetics of many drugs. The largest family of drug transporters is composed of the ATP-binding-cassette (ABC) transporters (6), which are involved in the cellular efflux of many clinically important drugs, including several antineoplastic agents. To date, ABC transporters have perhaps best been known for their role in tumor multidrug-resistance, and many comprehensive reviews have been written on this subject (7–10). The family of uptake solute carriers (SLC) also plays a central role in the cellular trafficking of numerous drugs, particularly the organic anions. Studies over the past decade have identified inflammation-mediated changes in the expression and activity of many of these drug transporters, which can significantly impact drug clearance. Hence, the present review encompasses information reported to-date on the regulation of several, key drug transporters under inflammatory conditions.
ATP-Binding Cassette (ABC) Drug Efflux Transporters
Found in all living organisms, ABC transporters are involved in almost every cellular, biological, and physiological system. Across many species, several thousand distinct ABC genes have been identified, all of them sharing a highly conserved domain: the ATP-binding cassette, also termed the nucleotide binding domain (NBD). Because the bulk of the ABC transporters are efflux pumps involved in the active transport of a wide variety of substrates (Figure 1⇓), the NBD acts as the site for substrate binding and consequent ATP hydrolysis, thus providing the energy required for transport (11). Conserved sequences specific to ABC transporters are found within the NBD, namely the Walker A and Walker B motifs, and the C (or ABC signature) motif. The large array of substrates that are transported by ABC transporters are a chemically diverse group, ranging from bulky lipophilic drugs and toxins, to organic anions, carbohydrates, and bile acids (12).
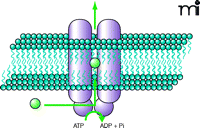
Illustration of an ABC drug-efflux transporter. An ABC drug transporter is typically composed of two substrate-specific membrane-spanning domains, which provide a pathway for solutes, and two substrate-binding cytosolic nucleotide binding domains which provide the site for ATP hydrolysis.
Although there are a large number of different ABC transporters, ABCB1, ABCC1–3, and ABCG2 are of particular importance, as they participate in the disposition of many clinically important drugs (13) (Table 1⇓). They are largely known for their involvement in the development of multidrug resistance, owing to their ability to export numerous anticancer drugs from tumor cells. As there are several excellent in-depth reviews available (11–18), we have provided only brief descriptions of these transporters (Table 2⇓).
Clinically Important Substrates of ABC Drug Efflux Transporters
Overview of Transporter Localization and Substrate Chemistry
Pertinent ABC Proteins in Drug Transport
ABCB1 (P-glycoprotein, Pgp), a 170-kDa membrane-bound transporter encoded by the multidrug resistance gene (MDR1 in humans and Mdr1a and Mdr1b in rodents) mediates the extrusion of a plethora of endogenous and exogenous xenobiotics. Although there is little structural or functional similarity between the known substrates, most are large, lipophilic, and either uncharged or cationic. Pgp is most highly expressed in the apical membrane of epithelial cells in the liver, kidney, intestine, colon, brain, testis, and placenta (19), suggesting that it plays a role in protecting tissues against potential toxins. Formerly considered a Pgp, ABCB4 (Mdr2), which is expressed exclusively in the apical membrane of hepatocytes, encodes a membrane glycoprotein exhibiting high sequence similarity to ABCB1. Although ABCB4 does not transport drugs, it does function as a physiologically important phospholipase. ABCB11, the bile salt export pump (BSEP), once termed the “sister of P-glycoprotein (SPGP)”, also shares high sequence identity with Pgp. It is localized almost exclusively to the apical membrane of hepatocytes but has also been detected in placenta (20). The role of ABCB11 in drug transport is limited.
ABCC1 (MRP1) was the first identified protein of the multidrug resistance-associated protein (MRP) family. MRP1 is a 190-kDa protein found in the basolateral membrane of epithelial cells of the gut, brain, kidney, lung, placenta, and testis. Very small amounts are found in liver. MRP1 is largely involved in the transport of amphipathic organic anions, lipophilic drugs, and glutathione-, sulfate-, or glucuronate-conjugated compounds (13). ABCC2 (MRP2) was initially termed the canalicular multispecific organic anion transporter (cMOAT) because it transported a broad range of lipophilic anionic compounds from hepatocytes into bile (21). The substrates transported by MRP2 are structurally diverse and include many large, lipophilic compounds. This 190-kDa transporter is expressed on the apical membrane of many tissues including the liver, kidney, intestine, brain, placenta, mammary cells, and gall bladder. Its localization in these tissues suggests that it plays a protective role in the body by promoting the excretion of potentially toxic xenobiotics. ABCC3 (MRP3) is a 190- to 200-kDa transporter that is most highly expressed in the basolateral membrane of liver, intestine, colon, and kidney, with expression also detected in the brain, pancreas, prostate, gall bladder, lung, and placenta. MRP3 serves as a diverse organic anion transporter with a high substrate similarity with MRP2, but has a greater affinity for glucuronide conjugates, whereas MRP2 preferentially transports glutathione conjugates (22). Therefore, MRP3 may be important as an alternative or compensatory route of transport in the absence of MRP2. There are several other members of the ABCC transporter family (ABCC4–C9); however, their roles in drug transport and inflammation have not been established.
Of the ABCG family of transporters, only ABCG2 is known to transport drugs. ABCG2, also known as the breast cancer resistance protein (BCRP), is a 72-kDa half-transporter which forms a functional homodimer (23). BCRP is most highly expressed in placenta, with expression also observed at the apical membrane of the liver, intestine, kidney, and brain. This expression pattern suggests that BCRP also plays an important role as a defensive barrier, limiting exposure of sanctuary sites to drugs or toxins. There is a great deal of substrate overlap between BCRP, Pgp, and some MRP transporters (Table 1⇑), although BCRP substrates do not exhibit the same extent of structural diversity as Pgp.
The Solute Carrier (Slc) Family Of Uptake Transporters
Cellular uptake is also an important determinant of drug absorption and bioavailability. The Solute Carrier (Slc) family of transporters comprises transporters that are expressed on the basolateral membrane and which mediate the uptake of substrates into cells such as hepatocytes. One of the larger groups of uptake transporters is the organic anion–transporting peptide (OATP, Slc21) family [reviewed in (24, 25)]. This is a family of basolateral transporters that mediate the sodium-independent uptake of compounds from the plasma. Their general protein structure is composed of twelve transmembrane domains with numerous sites for glycosylation and protein kinase C phosphorylation. Within the SLC family of drug transporters, only a few members have been examined in inflammatory conditions, and these are briefly described below (Table 2⇑).
Pertinent Slc Family Proteins in Drug Transport
The organic anion–transporting polypeptide 1 (OATP1, OATP1a1, Slc21a1) is an 80-kDa transporter predominantly expressed in the liver and, to a lesser extent, in kidney, brain, lung, and colon. OATP1 transports a broad spectrum of anionic drugs and substrates that are structurally unrelated, and contributes to drug absorption. OATP1 might also function as an anion exchanger, with the efflux of glutathione possibly being the driving force for substrate uptake (26). Organic anion-transporting polypeptide 2 (OATP2, OATP1a4, Slc21a5) is a 76-kDa bi-directional transporter that is highly expressed in the basolateral membrane of hepatocytes and has also been detected in kidney, brain, retina, heart, lung, spleen, skeletal muscle, and testis. OATP2 transports a broad range of substrates, and much like OATP1, OATP2 has a likely role as an anion exchanger. Sodium taurocholate co-transporting polypeptide (NTCP, Slc10a1) is a glycosylated, sodium-dependent transporter expressed exclusively at the basolateral membrane of hepatocytes. It preferentially mediates the sodium-dependent uptake of taurine- and glycine-conjugated bile salts, and is responsible for 80% of taurocholate uptake from the plasma. Although there are other important families of uptake transporters, such as the organic cation and anion transporters (Oat and Oct) or various nucleoside transporters, the expression or regulation of these has seldom been described in inflammation, and, thus, they are not included within this review.
The Acute Phase Response (APR)
Inflammation is the body’s defensive response to injury, tissue damage, or infection and involves many systemic and metabolic changes. The immediate reaction, termed an acute phase response (APR), is often associated with fever, vasodilation, leukocytosis, and metabolic changes. The APR usually lasts twenty-four to forty-eight hours and encompasses initial tissue injury resulting in cytokine release and receptor binding, activation of signaling cascades, followed by the synthesis and release of acute phase proteins. The end of the APR usually signals the restoration of homeostasis; however, failure of the APR to subside can develop into chronic inflammatory disease. Although chemicals such as nitric oxide, prostaglandins, leukotrienes, and histamine may contribute to the APR, cytokines are the principle mediators of inflammation. These small (< 40-kDa) soluble peptides are synthesized and released from various cell types, and their pattern of release is often specific to the inflammatory stimulant and the extent of tissue injury. The pro-inflammatory cytokines, including tumor necrosis factor (TNF)-α, interleukin-1 (IL-1), interleukin-6 (IL-6), and interferon (IFN), further stimulate the synthesis of other cytokines and chemokines that bind to receptors on epithelial membranes. These cytokine–receptor interactions trigger complex signaling cascades that lead to alterations in gene expression. The expression of several hepatic proteins––such as α1-acid glycoprotein, C-reactive protein, and α1-antitrypsin––is increased, resulting in plasma concentrations elevated as much as 200-fold, whereas the expression of several hepatic proteins––such as albumin, transferrin and many of the cytochromes P450––is decreased. The reason for the reduced expression of these latter proteins remains uncertain; however, there is speculation that they are mobilized as a source of nucleotides and amino acids for the synthesis of required acute phase proteins.
Modeling The Apr
Investigations of the APR in vivo commonly employ the well-characterized endotoxin lipopolysaccharide (LPS) or turpentine models. Intraperitoneal or intravenous administration of endotoxin, a major cell-wall component of Gram-negative bacteria, imposes a systemic inflammatory response characterized by symptoms such as fever, hypotension, and tachycardia. It is worthwhile to note that endotoxin from different bacterial strains effects differences in cytokine release and gene expression (27, 28). In general, however, administration of endotoxin results in an elevation of TNF-α, IL-1β, IL-6, and IFN-γ circulation in plasma (29). Knockout mice deficient in either IL-6 or IL-1β elicit a normal inflammatory response to endotoxin, although the secretion of TNF-α is significantly greater in IL-6–null mice than in IL-1β–null mice (30–33), suggesting the presence of compensatory or redundant mechanisms in the response to endotoxin in cytokine-deficient mice. On the other hand, the subcutaneous or intramuscular injection of turpentine, a chemical obtained from the distillation of resin from pine trees, induces a local, aseptic inflammatory response through the formation of a dermal abscess that subsequently triggers a systemic APR. Although multiple cytokines are thought to regulate the inflammatory response to endotoxin, the response to turpentine appears to be mainly dependent on the activity of IL-6, as IL-6−/− mice exposed to turpentine do not develop an APR (30, 34, 35). These studies clearly demonstrate the key involvement of cytokines in the APR and that the pattern of cytokine release is dependent on the inflammatory stimulant. Additionally, other models of inflammation that have been studied include transgenic or chemically-induced animal models of cholestasis, ulcerative colitis, inflammatory bowel disease, arthritis, and chronic renal failure.
Impact Of Organ Inflammation On Transporter Activity And Expression
Altered distribution of drug to sites of action or toxicity, and compromised secretion of drugs into bile or urine are major mechanisms by which inflammation may precipitate abnormal drug effects. To date, the vast majority of studies have examined the impact of inflammation on hepatic gene expression, as the liver is key to the acute inflammatory response and participates in the metabolism and biliary secretion of drugs. As discussed earlier, infection and inflammatory conditions mediate decreased expression of many P450 drug-metabolizing enzymes in liver, and many of the underlying molecular mechanisms have been delineated. Historically, however, the impact of inflammation on drug transporters had not been well established; thus, studies over the past decade have examined the interplay of inflammation and drug transporter regulation. More recently, studies have examined the implications of inflammation in other epithelial tissues. In particular, extrahepatic tissues of greatest interest to drug disposition research include the intestine, kidney, blood-brain barrier, and placenta. Expression of drug transporters in these tissues is critical to the processes of absorption, distribution, and excretion of its substrates.
Liver
There are several important drug transporters that are expressed in liver (Figure 2⇓). Of particular interest to hepatic-biliary drug clearances is the canalicular efflux transporter Pgp. Initial studies examining the impact of inflammation on this drug transporter demonstrated that turpentine-induced APR in rats elicited a 50–70% reduction in the in vivo hepatic expression and activity of Pgp within twenty-four to forty-eight hours after treatment (36). The reduction of Pgp expression and activity stemmed from suppression of Mdr1a and Mdr1b gene transcription. Endotoxin-induced inflammation also elicits significant reductions in the constitutive and induced expression of Pgp in rodents (37); however, divergent effects on transcription (as measured by mRNA accumulation) have been reported. Whereas the expression of Mdr1a mRNA is decreased in both endotoxin-treated mice and rats, the expression of Mdr1b mRNA is decreased in mice, yet increased in rats (38–41). As cytokine release is model-, strain-, and species-dependent, it was hypothesized that decreased expression likely occurs through a cytokine-mediated pathway. Indeed, studies with pro-inflammatory cytokines demonstrated that administration of IL-6 suppressed the expression of Mdr1/Pgp in vitro in primary rat hepatocytes and in vivo in mice, thereby suggesting involvement of IL-6 in the inflammation-mediated decrease of Pgp expression (42, 43). Decreased rates of Mdr1 gene transcription were also detected in IL-6–treated rat hepatocytes (42). IL-1β also contributed to Pgp suppression in rodents both in vivo and in vitro, whereas a pronounced induction of Mdr1b expression was observed in mice treated with TNF-α. On the other hand, Miyoshi et al. (44) reported significant endotoxin-mediated decreased expression of Pgp in wild type but not in TNF-α–/– mice, alternatively suggesting TNF-α may participate in endotoxin-induced decrements of hepatic Pgp expression. Differential effects of TNF-α and IL-6 on Mdr1 expression might be responsible for discrepancies seen with various models of inflammation; however, these studies collectively demonstrate the key involvement of cytokines in modulating transporter expression and activity during an APR.

Hepatic drug transporters whose expression is influenced by inflammation. P-glycoprotein (ABCB1), MDR2 (ABCB4), BSEP (ABCB11), MRP2 (ABCC2) and BCRP (ABCG2) are involved in the efflux of substrates into bile, whereas MRP3 (ABCC3) mediates basolateral efflux into plasma. Sinusoidal uptake transporters include NTCP (Slc10a1), OATP1 (Slc21a1) and OATP2 (Slc21a5), although OATP1 and OATP2 are also involved in substrate efflux.
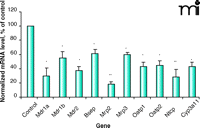
Effect of endotoxin on mouse mRNA. Representative graph showing the effect of endotoxin treatment on murine hepatic transporter mRNA expression. Results reproduced from (45, 55) (Mdr1a, Mdr1b, and Oatp1), and (60) (Mdr2, Bsep, Mrp2, Mrp3, Oatp2, Ntcp, Cyp3a11). Mice (n = 4–6), injected (ip) with saline (control) or 5 mg/kg endotoxin (LPS), sacrificed six hours post-treatment. Bars represent normalized mean values ± S.E.M., as a percentage of control value. ©2002, 2005. Used with permission by The American Society for Pharmacology and Experimental Therapeutics.
Pharmacokinetic studies in animal models confirm that functional changes in the expression and activity of Pgp, arising from systemic inflammatory conditions, result in corresponding changes to the in vivo transport, excretion, and tissue accumulation of model substrates. For example, endotoxin-treated mice exhibit significantly reduced biliary clearance of the Pgp substrate doxorubicin, which corresponds to decreases in hepatic Mdr1a/Pgp expression (45). Likewise, administration of the E. coli–derived Shiga-like toxin II impairs hepatobiliary transport of doxorubicin in rats, primarily because of decreased hepatic Pgp expression (46). Lower hepatic amounts of Mdr1a/Pgp in endotoxin-treated rats are also associated with a significant increase in the hepatic accumulation of the radio-pharmaceutical Pgp substrate 99mTc-sestamibi (41).
Several lines of evidence also suggest a cytokine-mediated decrease in MDR1/Pgp expression in human tissues. Reduced Pgp activity and MDR1 mRNA amounts are observed in IL-6- or IL-1β-treated human hepatoma cells. Diminished MDR1 gene expression and potentiation of chemosensitivity in human colon carcinoma cell lines incubated with IFN-γ, TNF-α, IL-2, or leukoregulin have also been reported (47–49). Consistent with the in vitro findings, observations of therapeutic synergism have been observed in patients given combinations of cytotoxic drugs with IFN or TNF (50, 51).
Another key canalicular transporter involved in the biliary secretion of drugs is MRP2. Over the past ten years, numerous research groups have reported dramatic reductions in the hepatic expression and activity of Mrp2 in several models of cholestasis, including endotoxemia (38, 52–54). The role of cytokines and other potential underlying mechanisms have since been examined. Studies in IL-6–deficient mice and in IL-6–treated wild-type mice suggest that IL-6 mediates decreased Mrp2 expression during inflammation (35, 55). Nakamura et al. (53) observed that IL-1α decreases Mrp2 expression in cultured rat hepatocytes, suggesting it is also involved in endotoxin-mediated suppression of Mrp2. Of note, intracellular redistribution of Mrp2 occurs during the early stages of cholestasis, with alterations in Mrp2 mRNA amounts occurring at later times (twenty-four hours post-treatment), which may mean that Mrp2 is regulated by both transcriptional and post-translational pathways (56). Indeed, dexamethasone treatment only partially impedes endotoxin-dependent decreases in Mrp2 expression (54, 57). Whereas Mrp2 levels are diminished in inflammation, several studies have reported compensatory increases in Mrp1 and Mrp3 in the livers of LPS-treated animals. Cytokine-induced expression of these transporters in human tissues is also suggested. For example, IL-6 treatment increases the expression and activity of MRP1 and MRP3 in human hepatoma cell lines (58). Interestingly, LPS incubated with rat or human liver slices (59) results in differential regulation of transporters, suggesting species difference between rats and humans. Amounts of rat Mrp2 mRNA are reduced twenty-four hours after LPS treatment, but amounts of the same transporter in humans, in contrast, are unaltered. However, human MRP2 protein is virtually non-existent twenty-four hours after LPS treatment, suggesting that a post-transcriptional process is involved in the regulation of this protein. A similar observation was made in the case of BSEP (59). Future studies utilizing human tissue samples will be crucial in elucidating the clinical implications of inflammation-mediated drug transporter changes.
Apart from the effects on Pgp or Mrp2, significant inflammation-mediated changes in the expression of many hepatic transporters have been reported. The amounts of mRNAs from several members of the ABC and Slc families of transporters are decreased in livers from in LPS-treated mice (60). Likewise, dramatic decreases in hepatic concentrations of Ntcp, Bsep, Mrp2, Mrp6, Oatps1, 2, 4, Oat3 and Oct1 were observed in LPS-treated rats (40). As this study found that treatment with the anti-inflammatory agent dexamethasone similarly attenuated changes in gene expression, it was proposed that regulatory responses were likely mediated through similar pathways. Studies to clarify the mechanisms by which these diverse transporters are regulated have been attempted. In vitro and in vivo results demonstrated a pronounced impact of endotoxin and cytokines on the regulation of hepatic organic anion transporters, whereas bile acid-induced cholestasis was found to only play a minor role (55). Similar to the downregulatory effects of IL-6 on Mdr1/Pgp, mice treated with various doses of IL-6 were found to have strongly decreased amounts of Mrp2, Mrp3 and Bsep, Oatp1, and Ntcp mRNAs (35). The effects of TNF-α and IL-1β were further uncovered in another study where mice treated with TNF-α had decreased amounts of Bsep, Oatp1, and Ntcp mRNAs, but mice treated with IL-1β also exhibited, in addition to reduced expression of the transporters mentioned above, decreased Mrp2 and Mrp3 mRNAs (61). LPS-treated rodents were found to have drastically reduced gene expression of the hepatocyte basolateral transporter Ntcp, likely modulated by IL-1β and TNF-α (62), but IL-6 involvement has also been suggested (63).
Currently, there is virtually no information reported on the impact of inflammation on Bcrp function in the liver. However, recent unpublished results from our group have demonstrated that Bcrp mRNA amounts are decreased in the livers of endotoxin-treated rats. The regulation of BCRP may be similar to that of Pgp; thus, inflammatory conditions might mediate a decreased BCRP expression, resulting in alterations in the disposition of its substrates. However, further experiments are needed to test this hypothesis.
Intestine
The intestine also influences the pharmacokinetics of drugs and limits the rate and extent of drug absorption and oral bioavailability. Inflammation of the intestine is not uncommon, and the APR in the intestine resembles that seen in the liver (64, 65). Indeed, IL-6 mRNA is increased in the intestine during inflammation. The in vivo expression of Mdr1a and Pgp, and to a lesser extent Mdr1b, is reduced in the jejunums of endotoxin-treated rats (66). Decreased Pgp has been associated with increased mucosal-to-serosal absorption of Pgp substrates across the intestinal sections of LPS-treated rats. Moreover, reductions in intestinal Pgp protein expression and activity are observed in rats with induced chronic renal failure (67, 68) and in the large intestine and colon of mice and rats with dextran sodium sulphate (DSS)-induced colitis (69, 70). Likewise, inflamed colonic and rectal mucosa samples from patients with ulcerative colitis reveal a strong reduction of Pgp expression (71). Increases in blood concentrations of the Pgp substrates cyclosporine and tacrolimus (FK-506) have also been reported in pediatric patients during diarrheal episodes (72).
Interestingly, a significant increase in Pgp expression and function was observed in the non-inflamed ileum of colitis-afflicted rats, alluding to the existence of a mechanism that compensates for the hindered Pgp activity in the colon (70). A similar increase in Pgp levels was reported for non-inflamed duodenal biopsies from children with Crohn’s Disease (73). Contrarily, in vivo and clinical studies of obstructive cholestasis found no significant difference in intestinal Pgp amounts when compared with controls (74). As with the liver, cytokines in the intestine likely mediate these effects. A cytokine-dependent reduction of MDR1 expression was observed in inflamed intestinal epithelium of patients with various gastrointestinal inflammatory disorders (75). IL-10–deficient mice spontaneously develop intestinal inflammation and are reported to have decreased Pgp function and expression all along the intestine (70). TNF-α or IL-2 reportedly suppresses Mdr1/Pgp expression and activity in vitro and in vivo in treated mice (76, 77). Furthermore, when plasma from rats with acute renal failure, which is associated with an inflammatory response (and presumably contains increased amounts of pro-inflammatory cytokines such as TNF-α and IL-6), was added to Caco-2 (human colorectal adenocarcinoma) cells in culture, reduced Pgp activity was observed in those cultured cells (78). However, Caco-2 cells treated with IL-1β and IL-6, exhibited increased amounts of MDR1 mRNA, whereas IFN-γ treatment of Caco-2 cells resulted in increased Pgp mRNA and protein expression but not activity, possibly owing to the modified localization of Pgp in the apical membrane after IFN-γ treatment (76, 79).
Decrements of other intestinal transporters have also been reported. Mrp2 is decreased in the intestine of LPS-treated rats and in several other models of inflammation, including cholestasis and chronic renal failure. In all cases, Mrp2 was dramatically decreased with respect to expression of mRNA (66), protein (74), or activity (68), pointing to a transcriptional or post-transcriptional regulation, possibly regulated by IL-1β (74). On the other hand, duodenal samples from patients with obstructive cholestasis exhibit a reduction of MRP2 protein, but not mRNA levels, suggesting post-transcriptional regulatory mechanisms are involved (74).
There have been few studies examining the impact of inflammation on BCRP. In patients with ulcerative colitis, colonic and rectal mucosa samples revealed that BCRP mRNA was strongly decreased during active inflammation (71). However, BCRP protein expression was not altered in duodenal samples from patients with obstructive cholestasis (74). Undoubtedly, additional studies are required in order to further demonstrate Bcrp regulation in episodes of inflammation.
Kidney
Although the kidney is crucial to the active and passive secretion of xenobiotics, to date the majority of studies have focused on exploring regulation of renal sodium, glucose, and urea transporters (80–82). Only a few studies have generated information regarding the regulation of renal drug transporters under inflammatory conditions. Endotoxin administration reportedly decreases the renal expression of Mdr1a and Mdr1b mRNA and imposes a significant reduction in the renal excretion of the Pgp substrate rhodamine-123 in rats (83). On the other hand, a threefold increase in the renal clearance of doxorubicin, with a corresponding induction of Pgp expression, has been observed in endotoxin-treated mice (45). These conflicting results likely stem from species differences in either Pgp regulation or inflammatory responses. Further studies are necessary to fully elucidate the inflammation-associated changes in the expression of renal drug transporters.
Brain
How are transporters regulated at the blood-brain border during inflammatory or infectious disease? Many studies have examined Pgp expression, as this protein is abundantly expressed in the several types of cells within the central nervous system (CNS) (11). Increased efficacy of several centrally acting Pgp substrates, including loperamide, fentanyl, and morphine, have been reported in a murine model of intestinal inflammation that results in overall suppression of Pgp (84), suggesting increased penetration through the blood-brain barrier. Increased intracranial accumulation of doxorubicin with a corresponding decrease of Pgp protein expression was observed in the brains of endotoxin-treated mice (85). In rats, systemic or CNS inflammation produced by intraperitoneal or intracranial LPS administration, respectively, also triggers marked decreases in Mdr1a mRNA expression in brain as well as increases in the brain concentrations of the Pgp substrates 99mTc-sestamibi and digoxin (39, 41). It is noteworthy that CNS inflammation results in a pronounced decrease of Oatp2 mRNA expression in the brain and liver of rats (39). In contrast, an increased brain:plasma concentration ratio of doxorubicin, along with induced Pgp expression, has been reported in mice treated with Shiga-like toxin (86). The distinct effects on Pgp expression may stem from alternate patterns of cytokine induction in these models. In vitro treatment of primary cultured rat astrocytes with gp120 (an HIV-1 viral envelope glycoprotein) leads to an inflammatory response that results in decreased Pgp expression and increased accumulation of Pgp substrates (87). This diminishment of Pgp is probably mediated by IL-6, as treatment of cells with IL-6, but not TNF-α or IL-1β, dramatically reduces Pgp expression (87). However, exposure to TNF-α caused isolated brain capillaries to undergo a specific signaling cascade that results in severely decreased Pgp-mediated transport, indicating a key role of TNF-α in Pgp regulation (88).
Placenta
Although pregnant women are subject to a number of inflammatory conditions, the expression and regulation of drug transporters in the placenta has only recently gained attention. Being the frontline barrier against xenobiotics and other harmful substances between the mother and the fetus, the integrity of the placenta is of utmost importance. Various placental ABC and other drug transporters have, of late, been identified (89–91). Systemic, endotoxin-induced inflammation in near-term pregnant mice reportedly imposes a dose-dependent decrease in the expression of several placental genes, including Mdr1a (92). Treatment with α-phenyl-N-t-butylnitrone (PBN, a reactive oxygen species) or with N-acetylcysteine (NAC, an antioxidant) attenuated the LPS-induced decrease of Mdr1a and of placental TNF-α, IL-1β, and IL-6, which were otherwise found in increased amounts after LPS treatment. Although the regulatory pathway was not confirmed, these findings suggest cytokine involvement. Likewise, a reduction in placental expression of Mdr1a and Mdr1b mRNA is seen in LPS-treated rats and is associated with in a pronounced increase in the fetal accumulation of the radiolabelled Pgp substrate 99mTc-sestamibi (41). The results together suggest that Pgp protects the fetus against uptake of exogenous compounds and that this protection might be compromised during maternal inflammatory conditions. Recent reports indicate decreased expression of Bcrp in the placentas of LPS-treated rats (93), and investigations are currently under way to elucidate the impact that this finding may have on the exposure of the fetus to Bcrp substrates. As the fetus could suffer grave consequences from compromised placental transport of xenobiotics, it will be essential to further study the role of inflammation on placental drug transporters.
Role Of Nuclear Receptors
Pro-inflammatory cytokines initiate many changes in gene expression by activating transcription factors, such as nuclear factor– κB (NF-κB) and NF-IL6 (also termed CCAAT/enhancer binding protein β [(C/EBPβ)]. More recently, nuclear hormone receptors have emerged as potentially important regulators of transporters during inflammation. Many transporters and drug metabolizing enzymes that are suppressed during inflammation, such as Mdr1, Mrp2, Mrp3, Bsep, Oatp2, and Cyp3A, are regulated by nuclear receptors such as the pregnane X receptor (PXR). The mRNA and protein expressions of PXR and other nuclear receptors are significantly decreased in the livers of LPS- or IL-6–treated mice (60). Of note, studies have demonstrated a significant reversal of Mrp2 diminishment by LPS and IL-6 in PXR-null mice, indicating that PXR is involved in the mechanism of Mrp2 suppression by these inflammatory stimuli. LPS treatment in wild-type mice also causes a marked decrease in PXR and Mdr1a in murine placenta (92), as well as in Mrp2 and Mrp3 in rat intestine (66). The expressions of farnesoid X-activated receptor (FXR), constitutive androstane receptor (CAR), and retinoid X receptor (RXR) are also reduced by LPS and by the pro-inflammatory cytokines IL-1β, IL-6, and TNF-α (94–99). Transactivation of the rat Mrp2 promoter by RXRα and by retinoic acid receptor α (RARα) was decreased by IL-1β treatment, resulting in the reduced expression of Mrp2 (100). Together, these findings suggest that changes in drug transporter expression during an inflammatory response may be mediated at least in part by changes in the expression of nuclear receptors.
Clinical Relevance
Differences in the bioavailability or efficacy of drug-transporter substrates, owing to an inflammatory response, occur in humans, although the mechanism behind these differences is generally unclear (Table 3⇓). Based on the reviewed findings of alterations in drug-transporter regulation (and thus, expression and functionality), however, it is likely that drug transporters have notable involvement. Elucidation of the impact of inflammation-induced changes in transporter expression on drug disposition is still in its infancy. Because ABC drug transporters such as P-glycoprotein, MRPs, and BCRP transport a large array of drugs, the effects of inflammation may drastically affect drug bioavailability or tissue uptake, and subsequently, drug efficacy or toxicity. Furthermore, it is possible that the severity of inflammation may alter the bioavailability of drugs indicated to treat conditions associated with inflammation. For example, protease inhibitors (HIV), anti-convulsants (epilepsy), anti-microbials (infection), and NSAIDS (arthritis) are all used to treat conditions with an inflammatory component, and many of these drugs are substrates for the drug transporters described. Diseases such as asthma, inflammatory bowel disease, epilepsy, cardiovascular disease, and cancer, as well as severe burns and organ transplants, which are linked with tissue damage or an immune response, may also affect drug disposition by altering transporter expression in the liver and other tissues. Future clinical investigations will be of vast importance in furthering our knowledge of human drug transporter regulatory mechanisms and inflammation-associated drug disposition.
Drug Transporter Substrates Affected by Inflammation in Humans
Conclusion
An ever increasing amount of data points to an interplay between drug transporters and metabolizing enzymes and to the involvement of key drug transporters in the phenomenon of inflammation-mediated changes in drug disposition. Although the in vitro and in vivo mRNA and protein amounts and activities of MDRs, MRPs, OATPs and several other classes of drug-transporting proteins are typically decreased during periods of inflammation, the exact mechanisms of regulation are still not fully explicated. Current opinion is centered on the involvement of pro-inflammatory cytokines such as IL-1β, IL-6, and TNFα, but nuclear hormone receptors also participate. During an inflammatory response, energy is focused on restoring homeostasis; hence, it is likely that any processes deemed noncritical, such as the manufacturing of drug transporters, may be temporarily shut down. However, much work remains to be done to advance our insight into the molecular mechanisms involved in the regulation of transporters during inflammation. Although hepatic regulation has been studied in some detail, there is limited information on the regulation of these transporters in other important tissues or organs. Furthermore, we have yet to establish the impact of inflammatory disease on the expression and activity of many other classes of drug transporters. There is also a substantial need for further clinical studies, as findings from animal or tissue models cannot fully be translated to humans.
Nonetheless, a growing number of examples of altered drug disposition resulting from the inhibition or induction of transporter activity through drug interactions, polymorphisms, or gene knockouts strongly indicate that uptake and clearance of numerous drugs could be compromised as a result of transporter modification. Clearly, the impact of changes in transporter expression by inflammation could have far-reaching consequences in terms of the pharmacokinetic and pharmacodynamic disposition of drugs. Continuation of investigative efforts towards understanding the mechanisms behind drug transporter regulation in inflammation is imperative.
- © American Society for Pharmacology and Experimental Theraputics 2007
References

Micheline Piquette-Miller, BSc Pharm, PhD, is an Associate Professor and Director of Pharmaceutical Sciences at the Faculty of Pharmacy, University of Toronto. She has been the recipient of numerous international and national research awards including the prestigious Leon Goldberg Young Investigator Award from the American Society of Clinical Pharmacology and Therapeutic (ASCPT). She is currently president of the Canadian Society of Clinical Pharmacology, and has served as Chair of Pharmacogenetics and Molecular Pharmacology and on the Board of Directors for ASCPT. Her research focuses on understanding inflammation-mediated regulation of drug transport proteins and its impact on drug disposition. E-mail m.piquette.miller{at}utoronto.ca; fax (416) 978-8511.

Shirley Teng, PhD, completed her graduate work in Dr. Piquette-Miller’s laboratory at the University of Toronto in 2006. She studied the role of PXR in the regulation of hepatic transporter expression under normal and inflammatory conditions.

Vanja Petrovic, BASc, is a biomedical engineer, currently pursuing a PhD degree at the University of Toronto, under Dr. Piquette-Miller’s supervision. Her studies concentrate on elucidating the effect of maternal inflammatory conditions on drug transporter regulation.

