Oncogenic Ras–Induced Expression of Cytokines: A New Target of Anti-Cancer Therapeutics
Abstract
The Ras family of small guanosine triphosphatases normally transmit signals from cell surface receptors to the interior of the cell. Stimulation of cell surface receptors leads to the activation of guanine exchange factors, which, in turn, convert Ras from an inactive GDP-bound state to an active GTP-bound state. However, in one third of human cancers, RAS is mutated and remains in the constitutively active GTP-bound state. In this oncogenic state, RAS activates a constellation of signaling that is known to promote tumorigenesis. One consequence of this oncogenic RAS signal in cancer cells is the upregulation of the cytokines interleukin (IL)-6, IL-8, and chemokine growth-regulated oncogene 1 (GRO-1). We review the evidence supporting a role for these cytokines in oncogenic RAS–driven solid tumors.

Ras and Cancer
The human RAS family of small guanosine triphosphatases (GTPases) consists of three members, encoded by NRAS, HRAS, and KRAS, that normally transmit signals from cell surface receptors to the interior of the cell. Stimulation of cell surface receptors leads to the activation of guanine exchange factors, which, in turn, convert RAS from an inactive GDP-bound state to an active GTP-bound state. In this active state, Ras adopts a conformation that permits effector proteins, such as phosphoinositide 3-kinase (PI3K), Raf, and Ral guanine exchange factors, to bind Ras, leading to their activation and propagation of signaling. Activation of RAS is terminated by GTPase-activating proteins (GAPs) that stimulate the GTPase activity of RAS, promoting hydrolysis of GTP and returning RAS to its inactive GDP-bound state (1).
In most cancers, RAS is inappropriately activated. In fact, 20–30% of all tumors harbor oncogenic point mutations in RAS (at Gly12, Gly13, or Gln61) that impair intrinsic and GAP-stimulated GTP hydrolysis, leaving oncogenic RAS in a constitutively active GTP-bound state (2). In pancreatic cancer, up to 90% of tumors carry an activating RAS mutation (2). If RAS itself is not mutated, often Ras signaling pathway components are inappropriately activated or repressed to promote Ras signaling. Specifically, upstream cell surface receptors that activate RAS, such as epidermal growth factor receptor and the closely related HER2/NEU, are often amplified or mutated to remain active, resulting in chronic RAS activation (1). In fewer cases, there is a loss of a RasGAP, keeping RAS in an active, GTP-bound state (1). Additionally, downstream activating mutations in Raf and PI3K or loss of the lipid phosphatase PTEN leads to activation of individual Ras effector pathways (1). Thus, the RAS pathway is inappropriately activated in most cancers.
A plethora of data demonstrates that constitutive activation of Ras promotes tumor phenotypes. Gain-of-function mutations to RAS that occur in human cancers endow the protein with the ability to promote cell proliferation, cell survival, cell migration, tumor growth, and metastasis in cell culture and animal models (3). In contrast, extinguishing oncogenic RAS expression in established tumors leads to tumor regression (4–6). Thus, understanding how RAS promotes tumorigenesis could have therapeutic value in the treatment of many types of cancers.
Oncogenic Ras and Cytokines
Activation of RAS induces an autocrine signal in tumor cells that promotes many tumor cell phenotypes (1), but it is now appreciated that RAS activation in tumor cells also leads to paracrine signaling to the tumor microenvironment (7–9). The tumor microenvironment plays a critical role in tumorigenesis (7–9). Modification of the tumor stroma, which is composed of fibroblasts, immune and inflammatory cells, adipocytes, and endothelial cells, is important for tumor growth and development. During tumor initiation, paracrine signaling from cancer cells modulates the surrounding stroma to establish a microenvironment conducive for tumor growth (9). Important modulations of the stroma include activation of fibroblasts and recruitment of inflammatory cells, which secrete proteases that release growth factors sequestered in the extracellular matrix (8, 9). The recruitment of endothelial cells for the formation of a vascular system, which provides oxygen and nutrients to the tumor cells (7), is also an important step in the establishment of a microenvironment. Infiltrating inflammatory cells in the tumor stroma also sustain and promote neovascularization (10). An increasing number of examples demonstrate that activation of RAS in tumor cells positively acts to foster changes in the tumor microenvironment. For example, oncogenic Ras has been shown to promote tumor cell invasion, in part due to the induction of matrix metalloproteinases that degrade the extracellular matrix, allowing tumor cells to escape and metastasize (11). Oncogenic RAS has also been linked to the induction of tumor angiogenesis. Loss of oncogenic RAS expression in tumor cells results in apoptosis of cells expressing CD31, a marker for endothelial cells, and a subsequent collapse in tumor vasculature and regression of the tumor, strongly suggesting that oncogenic RAS paracrine signaling promotes survival of endothelial cells (12). Additionally, oncogenic RAS has been shown to transcriptionally upregulate vascular endothelial growth factor (VEGF), which acts upon endothelial cells to promote angiogenesis (13, 14).
It has recently come to light that cytokines are one class of secreted proteins that could mediate oncogenic RAS paracrine signaling. Cytokines are small secreted proteins that act in a paracrine manner to facilitate the interaction and communication among cells, often eliciting an immune response. New findings reviewed here indicate that oncogenic Ras upregulates the expression of the cytokines interleukin-(IL)-8, IL-6, and chemokine growth-regulated oncogene 1 (GRO-1).
Sparmann and Bar-Sagi (15) reported that activation of a tetracycline-inducible oncogenic HRAS transgene in HeLa cells led to elevated mRNA and secreted protein levels of IL-8. Overexpression of oncogenic HRAS similarly increased expression and secretion of IL-8 in lung carcinoma (H125) and breast epithelial (MCF10A) cell lines, suggesting that this effect was not specific to one cell type. Increased IL-8 expression was critical for RAS-driven tumor growth in the HeLa cells, as antibody-mediated neutralization of IL-8 activity dramatically reduced subcutaneous tumor growth in immunocompromised mice (schematized in Figure 1 C⇓). Mechanistically, the tumor-promoting activity of secreted IL-8 was attributed to enhancing angiogenesis through paracrine signaling, a supposition that is supported by several observations. First, the tested HeLa cells lacked expression of the IL-8 receptors CXCR-1 and -2. Second, inhibition of IL-8 by injection of the neutralizing antibody greatly decreased the number of inflammatory cells within the tumor. Consistent with the link between inflammatory cells and angiogenesis, inhibition of IL-8 effectively halved the number of endothelial cells in early stage tumors. In addition, end stage tumors were clearly necrotic, consistent with the diminished tumor vasculature. Taken together, these data suggest that oncogenic HRAS–induced IL-8 expression in tumor cells plays an important role in tumor development by promoting the inflammatory response and tumor angiogenesis (Figure 2⇓). It is worth noting that IL-8 concentrations are elevated in the serum of cancer patients, including those with pancreatic (16), lung (17), melanoma (18), breast (23, 24), prostate (21, 22), and ovarian cancers (23, 24). Moreover, in breast cancer, elevated levels of circulating IL-8 correlate with advanced disease and diminished survival rate (19).
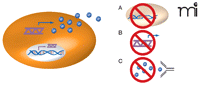
Inhibiting cytokine function impedes oncogenic Ras–driven tumor growth. Oncogenic Ras–mediated activation of cytokine gene transcription (double helix) in tumor cells (orange oval) leads to increased levels of cytokine mRNA (purple wavy line) and protein (blue spheres). The inhibition of cytokine activity may provide a means of treating Ras-driven cancers. Such inhibition has been effected by A) knockout of the cytokine-encoding gene; B) RNA interference (RNAi) targeting the cytokine mRNA; and C) anti-cytokine antibody–mediated neutralization. Each method of has been shown to inhibit Ras-dependent tumor growth. (See text for details.)
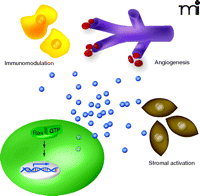
Oncogenic Ras–stimulated paracrine signaling promotes tumor growth. Oncogenic Ras (Ras-GTP) promotes the transcription of cytokine genes (double helix), leading to elevated levels of secreted cytokines (blue spheres) that act to modulate the immune system (yellow cells), promote angiogenesis (purple capillaries) and activate tumor stroma (brown cells).
To further elaborate the role of RAS-induced IL-8 secretion in tumorigenesis, Wislez et al. (25) utilized transgenic mice that have an oncogenic KRAS allele and consequently develop alveolar neoplastic lesions. In this model of lung cancer, mice that bear the oncogenic KRAS allele express elevated levels of two functional homologs of IL-8 [i.e., macrophage inflammatory protein 2 (MIP-2) and keratinocyte chemoattractant (KC)] relative to their wild-type littermates. Moreover, antibody raised against the CXCR2 receptor for the IL-8 homologs significantly reduced the number of lung lesions and malignancies (Figure 1 C⇑). Conversely, when isolated and grown in vitro, adenocarcinoma cells derived from the engineered mice were unaffected by the CXCR2-neutralizing antibody. The growth medium collected from these cells, however, elicits a positive response from alveolar inflammatory cells in a cell migration assay, and this cell migration is inhibited by the CXCR2-neutralizing antibody. Finally, progression of lung tumorigenesis in vivo correlated with the recruitment of CXCR2-expressing neutrophils and endothelial cells. Overall, these findings support a model whereby oncogenic KRAS–mediated secretion of IL-8 acts in a paracrine fashion to promote tumorigenesis.
In addition, GRO-1, belonging to the same cytokine family as IL-8, has been linked to RAS-mediated modulation of the tumor microenvironment. Specifically, immortalized human ovarian cells can be transformed by oncogenic HRAS or KRAS, which concomitantly results in the expression of GRO-1 and other cytokines (26). Yang et al. (27) found that short hairpin (sh)RNA-mediated knockdown of GRO-1 expression in such oncogenic HRAS–expressing (transformed) ovarian cells greatly reduced their tumorigenicity (Figure 1 B⇑). In this case, however, the paracrine effect was linked not to inflammatory cells, but rather to the fibroblast component of the tumor stroma. Specifically, treatment of cultured fibroblasts with recombinant GRO-1 promoted the growth arrest state of senescence. Moreover, the mixing of GRO-1–treated fibroblasts with immortalized but otherwise non-transformed ovarian epithelial cells prior to injection elicited tumors in immunocompromised mice. Lastly, cancerous human ovarian tissue samples contain greater numbers of growth-arrested fibroblasts than do normal control tissues, and the fibroblast cell lines isolated from these tumor tissues express higher levels of GRO-1. These results suggest that GRO-1 participates in RAS-induced tumorigenesis via the modulation of tumor-associated fibroblasts.
Our own studies have examined the role of IL-6 in oncogenic RAS–mediated cancer (28). These studies were prompted by the observation that IL-6 levels are elevated in the serum of patients diagnosed with cancers [pancreatic, lung, colorectal, melanoma, breast, prostate, and ovarian (29)] that are associated either with mutations in RAS or with mutations that affect RAS signaling (2). Moreover, IL-6 affects cancer phenotypes in several ways, including effects on cell survival (30), tumor angiogenesis (31–35), and perhaps tumor metastasis (36), often contributing to poor outcome and disease progression. We found that human embryonic kidney (HEK) cells that had been immortalized secreted elevated levels of IL-6 concentrations (relative to non-immortalized cells) and became tumorigenic upon induction of an oncogenic RAS construct (28), suggesting that IL-6 induction correlated with the tumorigenic activity of oncogenic RAS. A similar increase in IL-6 mRNA and protein was detected along with induction of tumor growth upon expressing oncogenic HRAS in human fibroblasts, myoblasts, and mammary epithelial cells. Using three independent shRNAs, we found that stable knockdown of IL-6 reduced the growth of tumors elicited by oncogenic HRAS-expressing HEK cells (Figure 1B⇑). Moreover, injection of mice harboring such tumors with a human IL-6–specific neutralizing antibody (which, like the shRNA, neutralizes tumor-derived IL-6 only) similarly reduced tumor growth (Figure 1C⇑). As with the detection of elevated IL-6 upon induction of the tumorigenic state by oncogenic Ras in different cell types, knockdown of IL-6 via shRNA similarly reduced tumorigenic growth of Ras-transformed myoblasts and fibroblasts. These results were validated in mice devoid of IL-6. Specifically, IL-6 knockout mice were significantly more resistant to the carcinogenic induction of skin papillomas that are typically characterized by oncogenic RAS mutations (37) (Figure 1A⇑). Moreover, knockdown of IL-6 in pancreatic cancer cell lines characterized by oncogenic KRAS also reduced their tumorigenic capacity. Mechanistically, oncogenic RAS–induced secretion of IL-6 appeared to promote tumor growth via paracrine signaling. First, cells in which IL-6 shRNA inhibited tumor growth lacked detectable IL-6 receptor. Second, shRNA-mediated reduction of IL-6 in these RAS-transformed cells did not inhibit their growth in tissue culture. Third, immunohistochemical analysis of tumors resulting from RAS-transformed HEK cells showed that knockdown of IL-6 reduced the number of endothelial cells in the tumor. As with the cytokines IL-8 and GRO-1, secretion of IL-6 is also induced by expression of oncogenic RAS in tumor cells, and this secretion promotes tumorigenesis in a paracrine fashion.
Targeting Ras-Induced Cytokines
Multiple models demonstrate that the inhibition of chronic activation of RAS greatly impedes the growth of tumors that arise either from activating mutations in RAS or illegitimate activation of cell surface receptors. Translating these observations to the clinic, however, has been difficult. For example, oncogenic activity of RAS is dependent on the posttranslational addition of lipid moieties (38), yet small-molecule inhibitors that specifically interfere with farnesyl transferase, the enzyme that adds one such lipid moiety, have failed to counteract oncogenic RAS and thereby provide anti-tumor activity. Moreover, these enzyme inhibitors fail to prevent KRAS and NRAS membrane targeting, because both of these Ras family members can acquire an alternative membrane-anchoring lipid moiety through the activity of geranylgeranyl transferase (39). Unfortunately, the use of inhibitors against geranylgeranyl transferase or both geranylgeranyl and farnesyl transferases is hampered in the clinic by toxicity (40).
Given the difficulty of directly inhibiting RAS, the inhibition of signals downstream of RAS has been pursued. The RAF inhibitor BAY 43–9006 has some efficacy in colon, breast, and non-small cell lung carcinomas (40), and small-molecule inhibitors of MEK1 and MEK2, downstream targets of RAF, have yielded positive results in the clinic in the treatment of renal cell carcinoma (40). Inhibition of the PI3K pathway by mTOR inhibitors has also yielded limited results (40). Thus, the concept of targeting RAS signaling has merit as a means to inhibit the growth of RAS-driven tumors.
Because their secretion is induced by oncogenic RAS, cytokines also represent an attractive target for the development of therapeutics that might interfere with RAS signaling. As discussed above, IL-6 and IL-8, as well as other cytokines, are elevated in the serum of patients diagnosed with cancers characterized by oncogenic RAS mutations (16–18, 20–24, 29), and oncogenic RAS–mediated tumorigenesis is significantly abated in the presence of neutralizing antibodies against such cytokines and by shRNA knockdown or ablation of cytokine-encoding genes (Figure 1⇑) (15, 25, 27, 28).
The targeting of cytokines may provide several clinical advantages. The inhibition of cytokines may be tolerated in patients, at least in the case of IL-6, as genetic ablation of IL-6 in mice is not lethal (41). In addition, cytokines are located in the interstitial space are thus much more bioavailable than intracellular targets. Indeed, antibodies that are already available or readily generated can be used to inhibit cytokine function, and antibody therapy is generally well tolerated in patients (42); preliminary studies already demonstrate anti-tumor activity of neutralizing antibodies against IL-8 and IL-6 (15, 28).
We suggest that humanized antibodies against cytokines like IL-8, GRO-1, and IL-6 may have utility in the treatment of Ras-driven cancers. Indeed, inhibition of proteins by humanized antibodies has proven successful in the clinic, as exemplified by bevacizumab (Avastin®) as well as a variety of other anti-VEGF drugs (43). Several completed and ongoing clinical trials have investigated the ability of monoclonal antibodies to inhibit IL-6 and thereby function as cancer therapeutics. Multiple myeloma in particular depends upon IL-6 and has been the subject of relevant antibody-based therapy (29), and additional trials are focusing on the treatment of prostate cancer with anti-IL-6 monoclonal antibody (44–46). The discovery that oncogenic RAS increases the secretion of cytokines that are important for tumor growth will indeed provide impetus for a number of strategies to target a central pathway that is activated in the vast majority of cancers.
- © American Society for Pharmacology and Experimental Theraputics 2008
References

Brooke B. Ancrile (right) is a graduate student at Duke University in the University Program in Genetics and Genomics. Her research focuses on the role of cytokines in Ras-induced tumorigenesis. Kevin M. O’Hayer (left) is a graduate student at Duke University in the Department of Pharmacology and Cancer Biology. His research focuses on the role of chemokines in Ras-induced tumorigenesis. Christopher M. Counter, PhD, (center) is Associate Professor at Duke University in the Department of Pharmacology and Cancer Biology and in the the Department of Radiation Oncology. His research focuses on cell transformation and immortalization. Address correspondence to CMC. E-mail count004{at}mc.duke.edu; fax 919–684–8958.

