Fragment-based ligand discovery
Abstract
Over the past five years, fragment-based ligand discovery has come of age. A number of compounds that evolved from fragments have entered the clinic, and the approach is increasingly accepted as an additional route to identifying new hit compounds in pharmaceutical discovery and inhibitor design. This review will summarize the current methods and ideas prevalent in the area. We will discuss the key concepts and advantages of fragment-based discovery, the approaches adopted in designing fragment libraries, the experimental methods that have been optimized for detecting fragment binding, and the strategies for evolving fragments to hit and lead compounds.
Introduction
The drug discovery process currently adopted by most pharmaceutical discovery organizations is to identify small organic compounds that modulate the activity of a particular biological target (usually a protein). This identification (or “hit”) provides compounds that are then explored in a so-called Hit-to-Leads process, according to which the activity of hit compounds can be improved by chemical modification and the corresponding analysis of structure–activity relationships (SAR). The Hits-to-Leads process thus generates a series of lead compounds that are then optimized with regard to the properties desirable for the development of candidate compounds for clinical evaluation.
The predominant method within large pharmaceutical companies for hit identification continues to be high-throughput screening (HTS), where large corporate collections of compounds (often many millions, augmented by parallel synthesis) are assayed for activity on a target. There continue to be improvements in HTS methods (1), but there remain issues with the approach. It can be a considerable challenge to configure an automated assay suitably robust to screen many hundreds of thousands of compounds for their binding properties. Despite considerable investments of time, many false positive hits (2) arise, which take further effort to triage and validate. In addition, the identification of hits is often restricted to compounds that bind with a KD on the order of 10 μM. To achieve an affinity at this level or better requires the hit compound to have sufficient complexity to match the requirements of the binding site. However, as elegantly analyzed by Hann et al. (3), increased molecular complexity (and size) reduces the probability of finding leads, because the decoration of compounds increases the chance that useful interactions will not be made by randomly chosen ligands (Figure 1⇓). It is not practically possible to overcome this limitation merely by synthesizing and testing more compounds. The number of potential compounds that can be synthesized rises exponentially with the number of atoms in the molecule, with estimates of 107 possible compounds for compounds containing twelve heavy (non-hydrogen) atoms (4) and 1060 compounds with up to thirty heavy atoms (5). Clearly, this number of molecules vastly exceed the practical demands of syntheses and assays, so the chance of finding a compound in a HTS library with the “right” combination of features is very low. It is therefore not surprising that many HTS screens fail to identify suitable hit compounds, particularly if compounds that inhibit a given class of target have not been studied previously. Finally, HTS requires substantial investment in automation, compounds, and library management facilities, which is not feasible for small pharmaceutical/biotechnology companies and most academic groups.

The task of high-throughput screening (HTS). The task of HTS is to identify compounds that bind to the target (left; schematized as an empty puzzle frame) from within the corporate compound library (shown in blue area). The right side of the figure shows an idealized hit compound binding to the target. Some of the compounds within the library contain moieties (each moiety represented as a single color) of the right shape and chemistry to bind to limited areas within the target. However, the compounds represented in the compound library in this case all contain additional groups that prevent binding. The schematic grossly simplifies ligand binding, but is meant to portray the preponderance of ineffective compounds within the chemical library.
Modern methods of structure-based discovery have provided three approaches that have had an impact on this overall process (6). The first exploits the structural assessments of hit or lead compounds bound to a target active site in order to improve drug-like properties. This approach could improve affinity or selectivity, or identify regions of the compound that can be modified to affect bioavailability and/or physico-chemical properties without affecting binding to the target [for an example, see (7)]. The second approach is virtual screening, where computational docking methods are used to identify which members of a large in silico library of accessible compounds might fit into the structure or define a pharmacophore for the target (8). Despite intensive development by the computational community over many decades, the Achilles heel of these in silico approaches is the inability to correctly calculate binding affinity and thus discriminate true from false positive hits. In general, these computational approaches are reasonably successful at predicting the conformation and position of ligands that are known to bind to a target site and can, with care, sometimes identify novel hits from a larger database of compounds; however, they usually fail to provide an accurate ranking of the binding affinities of ligands for a target (9).
The third innovation in structure-based methods is fragment-based lead discovery. Phil Hajduk has summarized the pioneering work from the Abbott group on the SAR by NMR approach that was the first method published in fragment-based ligand discovery (10). The past few years has seen publications from many other groups that have developed variants of the fragment approach and have successfully used the methods to generate clinical candidates [see (11–15) for review].
The aim of this review is to provide an overview of the current methods, practices, and concepts in fragment-based discovery. The central tenet is that the screening of small molecular fragments (typically containing less than eighteen heavy atoms and a molecular mass no greater than 250 Da) can reveal core templates that make key interactions with portions of a target binding site. Subsequent (almost exclusively structure-guided) elaboration of the fragment can then introduce interactions and compound features that provide the required affinity and selectivity, while conventional medicinal chemistry considerations optimize drug-like properties. Crucially, this incremental buildup allows researchers to sample a small library of hundreds of modular fragments that effectively represents the orders-of-magnitude greater chemical diversity that would have to be screened in a library of larger ligands. Although the fragment linking approach (see below) is a rather special case, the discovery of a potent inhibitor of the anti-apoptotic protein Bcl-XL using NMR and parallel synthesis (16) provides useful illustration of this idea. Discovery of this inhibitor was based on two fragments, binding separate target sites, that were identified from libraries of 10,000 and 3,500 compounds that could be combined by twenty-one different linkers. It would require, theoretically, the synthesis of 700 million compounds to produce all the possible combinations. Sequential screening of first 10,000 and then 3,500 fragments required only 13,500 experiments followed by up to twenty-one attempts at linking.
How Are Fragments Different?
Although any fragment that binds can be regarded as just a small, weak hit, establishing successful fragment-based discovery had depended on technical developments in three different areas: designing a fragment library, detecting which fragments bind to the target, and evolution of the fragments to larger hit compounds. The endeavor has brought about a major paradigm shift among medicinal chemists as they have come to realize that small, weakly binding fragments (often with mM binding affinity) could rapidly be optimized to result in lead compounds with nM affinities. Long before this realization, Jencks had established, over twenty-five years ago, an essential principle at the base of this new paradigm. He recognized that binding of a molecule has to overcome considerable translational and rotational entropy (17) and displace solvent before establishing an interaction with target. After the molecule (or fragment) has achieved an interaction, additional functionality can rapidly provide higher affinity. However, perhaps the most influential concept for the acceptance that fragments have value as starting points for optimization is that of ligand efficiency (18), which is defined as the binding energy (ΔG) per non-hydrogen-atom. Various retrospective studies have emphasized the importance of considering the amount of free energy of interaction gained by each heavy atom or functional group added to a compound (19) for successful hit and lead optimization campaigns.
Design of Fragment Libraries
One feature of the fragment approach that is attractive to both small companies and academic groups is the relatively small numbers of compounds that need to be maintained within a fragment library. However, as with any screening approach, the design of the library is critical to ensure that relevant hits of sufficiently high quality can be obtained. There are few publications that deal explicitly with fragment library design. The details of the methods and procedures particular to each project are, perhaps unsurprisingly, proprietary [but see the recent review of Hubbard et al. that summarizes what has been published (15)]. Nevertheless, some general principles can be identified, and a generic scheme for library design is shown in Figure 2⇓.

Generic design of a fragment library. See text for details.
Three general criteria, or “filters,” can be identified in the design of fragment libraries. The first criterion is that compounds have to meet the physico-chemical properties, (e.g., pertaining to solubility, size, or shape) that are dictated by the screening methodology. Second, the compounds should not contain or result in obviously toxic or reactive templates. Third, the fragments should have features suitable for evolution into larger, higher-affinity hit compounds. This third criterion is perhaps the most subjective to the experience (and prejudice) of the medicinal chemists who use the fragments. In some cases, each fragment is visually inspected before it is accepted into the library. Other libraries may be composed of compounds that have been derived from inspection of known drug-like molecules such as the SHAPES approach from Vertex (20) and the Drug Fragment Set at Astex (21). There are also varying approaches to designing a library based on target pharmacophores (22) or virtual screening to select libraries according to desired target classes such as kinases or phosphatases (21). Such target-derived filters usually increase the hit rate within the established library. However, the greatest advantage of the fragment approach remains the potential for identifying new chemotypes that unexpectedly satisfy the features of the target binding site, and many libraries do not have this step of selecting compounds to be biased towards a particular target class. In all cases, the members of a library must be chosen to give as diverse a representation of chemical space (however that may be measured) as possible.
It is possible to identify two main strategies under the overall umbrella of fragment-based discovery, where structure-guided methods are used to evolve weak-binding small molecules into larger higher-affinity compounds. One approach, often referred to as scaffold-based discovery, is to have a 10,000–20,000–member library of compounds with an average molecular weight of 300 Da. Compounds of this size can be screened for hints of activity in conventional binding or activity assays (23). In contrast, the standard fragment approach is to use a relatively small library, of 500–2000 compounds, with an average molecular weight between 110 and 250 Da. These typically bind to the target with a KD in the 100μM to 10mM range. In both cases, the detail of the binding of the hit fragments to the target is then characterized by crystallography (Figure 3⇓). The need to detect such weak interactions has encouraged the development and improvement of a battery of biophysical assay methods.
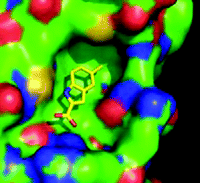
The visual power of crystallographic data. Shown here is the binding of a fragment to the protein Pin1. (See text for details.)
Detecting Fragment Binding
The main challenges for detection of weak binding are the sensitivity of the method, the solubility of the ligand, the stability of the protein, and interference from non-specific binding. Well established “tool compounds” are available for most classes of target. Examples of such tool compounds include: staurosporine for kinases; peptides chosen to partner in protein–protein interactions; and generic protease inhibitors. These tool compounds are valuable, as competitor ligands, for establishing optimal conditions for fragment binding and to confirm the specificity of hits. As with any HTS, the reliance on biophysical methods can result in frequent, non-specific hits (24, 25). However, the smaller size of the fragment library makes it feasible to check each compound with respect to solubility, self-aggregation, reactivity, or any other property that may underlie a false hit as the result of non-specific binding.
Conventional activity and binding assays, although generally rapid and amenable to automation, are generally not effective in the identification of ligands with KD values that are weaker than 500 μM. X-ray crystallography was one of the earliest method for demonstrating the nature of weak binding of ligands to proteins, as low-affinity components of the crystallization liquor could be detected on the basis of electron density. The binding, at high concentration, of very small fragments was explored in some detail by Ringe and others (26, 27), who were able to observe unique and specific weak binding by X-ray crystallography. However, the first published example of ligand discovery from fragments came from the Abbott group, who used NMR spectroscopy as the tool to detect binding and determine structures (11). The structural teams at Abbott also developed X-ray crystallography as a screening tool (28), but it was the small companies, such as Astex and SGX, who implemented (and promoted) the fragment discovery approach by crystallography (21, 29). This involves determining the crystal structure of protein soaked or co-crystallized with a mixture of fragments. It has the advantage that the binding mode of the fragment is immediately available, but it does requires a crystal form with an accessible binding site and (if soaking) which can withstand high concentrations of fragments. Crystal packing can occlude the binding site, requiring crystal engineering. In addition, a large investment is needed for data collection and interpretation if screening whole libraries. Although some groups still screen by crystallography, most practitioners now use a range of predominantly biophysics-based methods as a more cost- and time-effective way to screen for fragment binding and then confirm binding of hits by X-ray crystallography.
There are two main ways in which NMR spectroscopy can be used to observe ligand binding, differentiated by whether binding is monitored through changes in target or ligand signals. With some exceptions, the ligand monitoring approach is more widely used for screening fragment libraries, as it has fewer demands on instrument time, does not require isotopically labelled protein, and can, in principle, work for any size of molecular target. In addition, this approach addresses the physical state of both protein and ligand, allowing protein and ligand solubility, aggregation, and degradation to be checked at each screen. In contrast, protein monitoring experiments, such as heteronuclear single quantum correlation (HSQC) experiments, are useful for direct investigation of the target binding site. Figure 4⇓ shows an HSQC spectrum in which each NMR peak corresponds to the chemical environment of a given nitrogen atom within the protein; because the binding of a compound to the target changes the chemical environment in the binding site of the protein, this change will be reported in specific peaks in the HSQC spectrum.
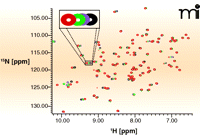
15N NMR—HSQC measurements. Each peak in the NMR experiment reports on the local chemical environment of a nitrogen atom; this environment changes during compound binding and is reported as shifts in peaks. The method requires large quantities of 15N-labeled protein that must be stable at 100 μM in low-salt buffer for many days. Here, a representative HSQC spectrum of the protein Pin1 with detail showing how peak positions shift (schematized by changes in color of peak) as concentration of a ligand is increased (J. Murray, Vernalis, unpublished results).
In the past five years, surface plasmon resonance (SPR) has gained increased popularity for this initial screen of fragment binding (30). Here, the binding of a fragment to the protein is observed as a change in the mass of the molecular system attached to a chip surface (Figure 5⇓). If the protein itself is attached to the surface (25), the measurement is direct and can provide kinetics of binding and a KD value. Alternatively, small-molecule ligands attached to the surface (31) allow one to determine the concentration at which a fragment prevents protein binding to the attached ligand (i.e., determination of KI alone). The major challenge in SPR is to the robust immobilization (of either target or fragment) to the chip surface without affecting binding properties. If appropriate immobilization can be achieved, SPR is the technique of choice as it uses relatively small amounts of protein. The other biophysical technique which can provide KD-values and other thermodynamic parameters of ligand binding is isothermal titration calorimetry (ITC) (32). The quantities of protein and ligand required for such analysis are generally prohibitive for use in the screening phase, but there continue to be advances in miniaturization and the technique can be useful in validation and for the selection of fragments for evolution (discussed below). Data from an ITC experiment are presented in Figure 6⇓.
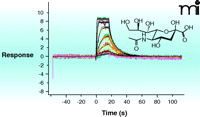
Surface plasmon resonance (SPR). SPR directly measure the change in the mass of a molecule attached to a surface as it binds to a second molecule from solution. SPR can routinely measure KD as weak as 5 mM. The method can be automated for relatively high throughput (hundreds of fragments per day), and can provide binding thermodynamics and kinetics. The challenge of the method is to fix molecules for assessment to the surface of a chip without interfering with possible interactions with molecules from the solvent. (See text for details.) The experiment shown here involves the titration of the compound Neu5Ac from solvent to a substrate-binding protein (SiaP) that had been covalently attached to the sensor surface.
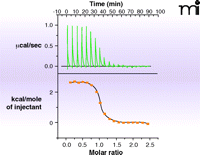
An example of isothermal titration calorimetry (ITC). The heat evolved or absorbed as a compound binds to a protein is a calorimetric property. ITC is limited by high solubility requirements for ligand and protein and by the amount of heat generated, but typically can measure KD values in the range of 10nM to 500μM. Shown in the data here is the titration of the tripeptide K-cyclohexylalanine-K to the protein OppA. The upper curve is the direct measurement of heat generated as ligand is added to protein; the bottom curve plots these values to provide a KD, which combined with the total heat generated (area under the curve) allows enthalpy and entropy to be estimated.
Another relatively new method is the fluorescence-based thermal shift assay (33). The temperature at which a protein unfolds, both in the presence and absence of ligand, is revealed by an environmentally sensitive fluorescent dye (34). This approach is economical, requiring large investments in neither reagents nor equipment. There has also been exploration of mass spectrometry (MS) methods for monitoring fragment binding (35). The technique has been of little use in monitoring non-covalent ligand interactions but can be a rapid way of identifying binding in cases where fragments bind in a covalent manner, such as the fragment-tethering approach developed by scientists at Sunesis. This strategy depends on the use of a thiol-containing fragment and a suitable cysteine residue (which can be engineered into a protein target) at the target binding site that may form a disulfide linkage. Subsequent elaboration of the fragment can include or exclude the covalent disulfide chemistry. A particularly elegant example combines this idea with dynamic combinatorial chemistry (35).
In general, it has proven difficult to configure robust enzyme or binding assays that require high concentrations of fragments for the determination of binding affinities. As large pharmaceutical companies begin to introduce fragment methods into HTS, additional effort to circumvent the problems of high-concentration screening is likely. To date, the published successes of this assay approach has been confined to higher-molecular weight scaffolds (23, 36).
A Typical Fragment Based Discovery Platform
Increasingly, most practitioners are converging on a similar approach to fragment-based discovery. Figure 7⇓ is a schematic describing the overall process implemented and developed over the past eight years at Vernalis (37). A relatively high-throughput method is used to detect fragment binding and then determine crystal structures of fragments bound to the protein target. For some targets, particularly, those that involve protein–protein interactions, it can be challenging to determine multiple crystal structures, and an additional validation step is then needed to characterize fragment binding. Assessment of protein–protein interactions, among others, is prone to artifacts, owing to non-specific binding to large, often hydrophobic, target surfaces.
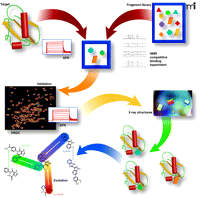
Overview of fragment-based discovery. The following description is based on processes developed and implemented at Vernalis (22, 44). A 1,200-member fragment library is screened in mixtures of up to 12 compounds for binding to the target protein by either NMR or SPR. Three NMR experiments are run (STD, CPMG, and LOGSY). A hit is defined as a fragment for which binding signals in all three experiments are reduced or abolished on addition of a competitive ligand. Alternatively, SPR is used where a robust attachment of the protein can be made to the sensor chip surface. Typically, a screen of 1,200 fragments results in between 10–100 fragment hits. Validation by HSQC and/or SPR is implemented for challenging protein–protein interaction targets, where it is necessary to use complementary biophysical measurements. Attempts are then made to determine the crystal structures of as many fragments bound to the target as possible, either by soaking apo-crystals or by co-crystallization. The resulting crystal structures define the position, binding mode, and interactions made by fragments. This information can then be integrated into structure-guided medicinal chemistry to evolve the fragments into lead compounds.
Integrating Fragments with Medicinal Chemistry
Figure 8⇓ schematically summarizes the different approaches that have successfully been used to optimize fragments into lead compounds, and an example of fragment evolution is depicted in Figure 7⇑. As discussed earlier, the idea of ligand efficiency has become an important concept to guide effective optimization of fragments into hits and leads, providing a quantitative measure for judging the effect of additional groups and moieties to the lead compound. Indeed, such quantitative analysis has become a hallmark of fragment-based discovery programs that seek to explain whether increased affinity during compound evoloution has not just come from adding non-specific or redundant bulk. [For example, see (13).]
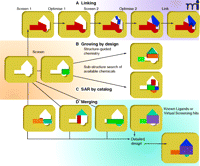
Evolving fragments into leads. The four main strategic categories are shown for evolving fragments into lead compounds.
The SAR-by-NMR approach is suited to targets that have relatively discrete binding pockets. There are a number of examples [primarily from the Abbott group (16)] where the linking together of sequentially identified and optimized fragments has generated potent inhibitors. However, most practitioners have found it more difficult to identify methods of chemical linking that preserve the binding and orientation of the fragments (38, 39).
One of the basic tenets of fragment-based discovery and development is that of fragment evolution, or fragment growth. Structural information about a core fragment bound to the target site can be used either to direct chemical synthesis (40) or to select from available compounds that contain the fragment as a sub-structure (so-called “SAR by catalog”). More ambitious is the idea of fragment merging, as in the evolution schema shown in Figure 7⇑, where the structure of different fragments, literature compounds, or virtual screening hits can be analyzed to design new compounds that incorporate features from various compounds [see (6)]. Although this idea of merging chemical features has been a hallmark of conventional medicinal chemistry, the fragment-based methods provide a richer tableau of options, and structural analysis of target–ligand interactions over the course of compound evolution provides confidence to embark on new chemistries.
Successful application of all these methods relies on close collaboration and communication among structural scientists, molecular modelers, and medicinal chemists. Successful partnership is perhaps the major requirement in successful optimization of fragments to leads. Indeed, a major reason for the success of small startup companies in developing and applying fragment-based methodologies may well stem from organizational emphasis on interdisciplinary approaches and the cross-fertilization of ideas.
Conclusions: What's Next FOR Fragments?
There are now many examples where fragment-based methods have provided the inspiration and guidance for the design of novel compounds (12, 13). These successes, particularly the potential to provide new chemical matter as starting points against novel target classes, has led to rapid take-up and deployment of the methods within large pharma—often alongside and providing complementary results to larger HTS campaigns.
So, where next for the methods? In many ways, the identification of fragments that bind to targets is generally a solved problem. However, it is possible to identify four main areas that require further development and innovation.
Fragment evolution in the absence of crystal structures.
A hypothesis that relates modifications of a compound to alteration of biological activity at a target (i.e., SAR) is essential; however, it is very difficult to measure activity for weakly binding fragments. It is probable that experience and improved practice in configuring robust assays based on methods such as ligand-observed NMR, SPR and thermal denaturation will provide more routine know-how for gathering SAR data for weakly binding fragments. Nevertheless, most practitioners continue to rely on information from the crystal structure of the fragment bound to the protein to provide ideas on how to evolve a fragment to a larger compound that is then on the scale of a reliable assay. Computational methods such as docking can provide some guidance when a suitable structure is available (15), but multiple crystal structure determination is to be preferred to identify subtle conformational changes which can confound the calculations.
Fragments for membrane-bound targets.
For the very many targets that are integral membrane proteins, practitioners face the challenge of configuring a binding assay in the presence of suitable solvent systems or membrane components. There can often be a delicate balance between assay components that maintain the structure and functionality of integral membrane protein targets and components that cause the assay to become overwhelmed by the non-specific binding of fragments to solvent systems or membranous elements. There are (as yet unpublished) reports of attempts to develop NMR or SPR based approaches in this area.
Fragment libraries.
Current databases of chemicals have been extensively mined for fragments, and there are an increasing number of fragment libraries from commercial suppliers. Nevertheless, the scope of available libraries could be widened by adding fragments that represent new chemistries. Fragment libraries could in particular benefit from diversity-oriented synthesis based on natural-product-derived cores (41) and scaffolds suitable for analysis of protein–protein interaction classes (42).
Decision making and the progression of fragments into “hits and leads.”
Most practitioners find that screening a suitably configured 1000-member library with a robust assay identifies 10 to 100 fragments. The challenge then is to decide which fragments to take forward in the attempt to evolve hits and subsequent leads. To date, decisions have been based on chemical tractability and information available from fragment–target crystal structures. There is a need for improved tools for visualization and analysis of multiple protein–ligand complexes and how to combine functionality into synthetically tractable compounds. Current drug discovery campaigns already include assessment of physico-chemical properties that can routinely indicate which fragments are the best candidates for development. These predictions are informed by consideration of the on and off rates for binding (43), provided for example by SPR methods, and whether binding is driven primarily by enthalpy or entropy, which can be provided from ITC measurements (44).
The last five years have seen real examples of fragment-based methods having an impact on the ligand discovery process for some classes of targets. The central feature is effective integration of structural information, modeling, and chemistry. This integrated approach can convert fragments into chemically attractive lead compounds against even quite challenging targets, allowing conventional structure-guided medicinal chemistry to be applied to identify clinical candidates. It remains to be seen how many of these candidates will survive the rigors of clinical trials. However, it is probable that the next five years will see more than one example of a compound, which began life as a fragment, successfully completing Phase II, and we will finally be in the era of fragment-based drug discovery.

Acknowledgments
We are extremely grateful to many colleagues at Vernalis and York for their work in developing the ideas of fragment-based discovery. In particular at Vernalis, James Murray, Ben Davis, Heather Simmonite, and Lee Walmsley provided some of the experimental data presented here and Ijen Chen and Nicolas Baurin contributed to the cheminformatics analyses. At York, Kerrin Bright, Yasuhiko Kanda and Michele Schulz provided both experimental data and useful discussions. M.F. is supported by a Wild Fund and BBSRC PhD studentship.
- © American Society for Pharmacology and Experimental Theraputics 2009
References

Marcus Fischer, MSc, is working toward his PhD with a focus on structural biology and virtual screening. He is currently investigating ligand binding to a set of carbohydrate-binding proteins using crystallography, NMR, SPR, ITC, and computational methods.

Roderick E. Hubbard, PhD, has spent his academic career at the University of York. During the 1980s, he developed molecular graphics and modeling systems for studying protein structure (HYDRA and QUANTA). He also helped to build (and directed) the Structural Biology Laboratory at York, now a major center for studying the structure and function of proteins. His research interests focus on structure, mechanism, and function in various protein systems and protein–ligand interactions. Since 2001, he has spent some of his time at Vernalis, where he helped establish and apply structure-based drug discovery methods. E-mail rod{at}ysbl.york.ac.uk; fax +44 1904 328266.

