Anti-HIV-1 Therapeutics: From FDA-approved Drugs to Hypothetical Future Targets
Human immunodeficiency virus type 1 (HIV-1) is the causative agent of the global AIDS pandemic. An intensive research effort over the past twenty years has led to the development of several classes of effective anti-HIV-1 drugs that have significantly improved patient survival (1). The acquisition of drug resistance, however, has limited treatment options for many patients, driving research to identify new drugs targeting novel steps in the HIV-1 replication cycle (2, 3) (Figure 1⇓). Recent advances in our understanding of HIV-1 replication at the molecular level may offer many new possibilities for therapeutic intervention.
The majority of clinically approved antiretroviral drugs target the viral enzymes reverse transcriptase (RT) and protease (PR) (Table 1⇓). RT inhibitors fall into one of two classes, based on their mode of action: the nucleoside-analog RT inhibitors (NRTIs), which are incorporated into the growing DNA strand and serve as chain terminators; and the non-nucleoside RT inhibitors (NNRTIs), which bind RT and induce conformational changes that inhibit RT polymerization activity (4). The RT enzyme also has intrinsic ribonuclease H (RNase H) activity, which degrades the RNA strand of RNA-DNA duplexes formed during DNA polymerization (5). At this time, there are no RNaseH inhibitors in clinical use; however, this is currently an active area of investigation (6). Protease inhibitors (PIs) competitively inhibit PR enzymatic function by binding to the active site of the enzyme (7). Inhibition of PR function prevents all proteolytic cleavage events associated with particle maturation, resulting in the formation of non-infectious, immature virus particles.
In 2007, raltegravir, the first drug targeting the viral integrase (IN), was approved for clinical use (8). A second compound, elvitegravir, is currently performing well in clinical trials (8, 9). Both raltegravir and elvitegravir act by binding directly to a DNA-IN intermediate formed during the integration of the viral DNA into the host cell genome, blocking the integration process and preventing the establishment of an infection (2, 8). An alternative but still hypothetical strategy for inhibiting DNA integration is to block the interaction between IN and the host cell factor lens epithelium-derived growth factor/transcriptional co-activator 75 (LEDGF). This virus-host interaction plays an important role in the integration process by tethering IN to chromatin (10). LEDGF contains a C-terminal IN-binding domain and an N-terminal sequence that interacts with chromatin (11, 12). The discovery of small molecules that block the interaction between IN and LEDGF could lead to a new class of integration inhibitors. Indeed, a dominant-negative, IN-binding fragment of LEDGF potently inhibits HIV-1 integration in cell culture (13, 14). A challenge that will be faced in developing LEDGF-based inhibitors derives from the fact that siRNA depletion studies have demonstrated that very low levels of this protein are sufficient to promote HIV-1 integration (12).
Two inhibitors of fusion and entry are currently in clinical use. T20, approved in 2003, is a peptide that blocks a critical conformational change in gp41 that is required for completion of the membrane fusion reaction (15). Limitations associated with T20 are the expense of peptide manufacture and its subcutaneous mode of delivery (16). Maraviroc, approved in 2007, inhibits the interaction between gp120 and the chemokine (C-C motif) receptor 5 (CCR5) coreceptor (15). A second CCR5 inhibitor, vicriviroc, is currently undergoing Phase III clinical trials (9, 15). Maraviroc is the first approved antiretroviral drug that specifically targets a host cell factor (CCR5) rather than a viral factor (17). By specifically targeting CCR5, rather than the other major HIV-1 coreceptor CXCR4, maraviroc use is restricted to patients infected with CCR5- but not CXCR4-tropic1 strains of HIV-1 (Box 1) (9). Despite this limitation, an intensive research effort to develop additional entry inhibitors is underway, as these drugs are potentially useful not only for treatment but also for prevention of HIV-1 infection (18).
Co-receptor Involvement in HIV-1 Infection
HIV-1 infection typically requires expression of a coreceptor, CCR5 or CXCR4 (15). CCR5 is expressed on primary macrophages whereas CXCR4 is abundant in T cell lines. Primary lymphocytes usually express both CCR5 and CXCR4. CCR5-tropic strains of HIV-1 tend to predominate early in infection and are generally less pathogenic than CXCR4-tropic strains. In approximately 50% of infected individuals, a switch to CXCR4 use occurs late in infection. This switch in tropism is often accompanied by an increase in viral loads and a decline in the numbers of CD4+ T cells in the circulation. Approximately 1% of the Caucasian population harbors a homozygous deletion in the CCR5 gene that markedly reduces susceptibility to HIV-1 infection (43).
The development of maturation inhibitors represents a novel and exciting breakthrough in antiretroviral drug research. This class is defined by compounds that target Gag to prevent PR-mediated processing at specific Gag cleavage sites or that bind CA to block core formation. Bevirimat, the first maturation inhibitor to be tested in clinical trials, operates by the first mechanism; it prevents the cleavage of a small spacer peptide (SP1) from the C-terminus of the CA domain, thereby blocking the formation of normal conical cores and severely disrupting virus infectivity (19–21). Bevirimat, however, does not impair PR-mediated cleavage of Gag monomers (19), implying that the binding site for bevirimat is created during the Gag assembly process. Learning more about the structure of the bevirimat binding site will be useful in developing additional compounds that disrupt CA-SP1 processing. Virion maturation can also be inhibited by blocking the CA-CA interactions required for core assembly. Two molecules, N-(3-chloro-4-methylphenyl)-N’-{2-[({5-[(dimethylamino)-methyl]-2-furyl}-methyl)-sulfanyl]ethyl}-urea) (CAP-1) and CA assembly inhibitor (CAI), have been identified that inhibit CA-CA interaction; these inhibitors bind directly to different regions of CA (22, 23). The CA protein is composed of two independently folded domains, an N-terminal (CANTD) and a C-terminal (CACTD) domain. Formation of the viral core following cleavage of CA from the Gag precursor involves the assembly of CA into a curved hexameric lattice; the inclusion of pentamers in the hexagonal array closes off the ends to form a structure that resembles that of fullerene cones assembled from elemental carbon (24, 25). The generation of this conical structure, which again is absolutely required for virion infectivity, requires several intra- and inter-hexamer interactions (26). CAP-1 binds a pocket at the base of the CANTD structure by an “induced-fit” mechanism; the aromatic ring of a buried Phe residue is displaced to create the CAP-1 binding site (27). CAP-1 binding appears to disrupt a critical CANTD-CACTD interaction. CAI is a peptide that binds to the CACTD and interferes with both CANTD-CACTD and CACTD-CACTD interactions (23, 28). Although CAP-1 and CAI represent mechanistically interesting leads, they are not drug candidates, in part owing to their weak CA-binding affinity. CAI has the additional problem of being cell-impermeable; however, chemical modification (i.e., “hydrocarbon stapling”) of CAI results in a cyclic, cell-penetrating derivative, NYAD-1, that displays antiviral activity in cell culture (29).
HIV-1 buds from cells by engaging the complex cellular endosomal sorting machinery (referred to as “ESCRT”) that normally acts to form multivesicular bodies. Direct interactions are required between small motifs in the Gag protein (known as “late domains”) and the cellular proteins Tsg101 and Alix (30, 31). The Gag–Tsg101 interaction, which is particularly important for virus release, involves a Pro-Thr-Ala-Pro (PTAP) motif in Gag and a bifurcated binding groove in Tsg101. The structure of the PTAP–Tsg101 binding site is quite similar to that of the interface between Pro-X-X-Pro motifs and their binding sites found on Src homology 3 (SH3) domains (32). Overexpression of the Gag-binding fragment of Tsg101 blocks the budding of HIV-1 and its relative, the feline immunodeficiency virus (33, 34). Several groups, including our own, are currently poised to initiate high-throughput screens for inhibitors of Gag-Tsg101 interaction. Such an approach may lead to the identification of small-molecule budding inhibitors and add to the arsenal of currently available antiretroviral drugs (35).
HIV-1 exploits a wide array of host factors and cellular machinery throughout the course of its replication cycle. Not all host cell factors, however, benefit the virus. In fact, cells from humans and other mammals have adopted strategies aimed at restricting retroviral infection (Figure 1⇓, molecules in red). Three such restriction factors that significantly impact HIV-1 biology have been described recently (36). First, the tripartite motif-containing protein TRIM5α interacts with the viral CA protein present on incoming reverse transcription complexes and leads to their premature degradation (37). Although HIV-1 infection is restricted by ectopic expression of TRIM5α from some species of non-human primates, HIV-1 is largely insensitive to the effects of human TRIM5α (37). Second, the cytosine deaminase APOBEC3G (apolipoprotein B mRNA-editing enzyme, catalytic polypeptide-like 3G) is incorporated into HIV-1 particles during virus assembly and in the course of reverse transcription in the next round of infection converts cytosines to uracils (38). This deamination leads to frequent guanine-to-adenine mutations in the newly synthesized viral DNA and to severely decreased infectivity. In response, HIV-1 evolved an accessory protein known as Vif (viral infectivity factor) that counteracts APOBEC3G by inducing its proteasomal degradation (39). Third, many human cell types express the interferon-inducible protein BST-2 (also known as CD317 or tetherin), which tethers budded virion particles, retaining them on the cell surface and preventing their release (Figure 1⇓, late events). The HIV-1 accessory protein Vpu, however, promotes particle release by counteracting BST-2 (40, 41). Thus, each of these host-cell defense mechanisms may present opportunities for therapeutic intervention. For example, a high-throughput screen for small molecules that block the ability of Vif to promote APOBEC3G degradation was recently reported (42). Although the mechanism of action of the most potent lead (RN18) from this screen remains to be defined, it apparently reduces the amount of Vif in an APOBEC3G-dependent manner, without stimulating overall proteasome-mediated protein degradation (42). Molecules with this activity could in theory be used therapeutically to tip the balance in favor of the host by preventing Vif from inducing the degradation of APOBEC3G. A greater understanding of the molecular basis for these host-cell defenses and HIV-1 countermeasures will increase the likelihood that useful antiviral strategies will be aimed at these host-pathogen interactions.
Notable success has been achieved in developing HIV-1-specific drugs that are clinically effective; however, because current drug regimens control but do not eradicate HIV-1 in the infected patient, long-term treatment is a necessity. This unavoidably leads to problems of drug tolerance and resistance. It is therefore clear that the field of HIV research must remain actively engaged in developing new drugs against novel targets. Recent and ongoing studies have provided an unprecedented amount of information on the molecular details of HIV-1 replication, thus revealing a number of new opportunities for pharmacological intervention. In our opinion, maturation inhibitors hold particular promise. Gag must be cleaved at five major processing sites by PR for maturation to occur; each of these sites therefore represents a potential target for drug development. Furthermore, the critical importance of the CA assembly pathway in core formation, and the existence of several CA-CA interfaces that are required for this process, present at least several additional target “hot spots.” The Vif-APOBEC3G axis also represents an attractive target that is only beginning to be explored. Although developing these targets into effective antiretroviral drugs will likely be a slow and difficult process, success in this effort will benefit the fight against HIV/AIDS.
Classes of Anti-HIV-1 Therapeutics: From FDA-approved Drugs to Hypothetical Future Targets
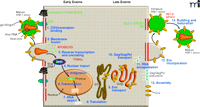
The HIV-1 replication cycle. Major steps of HIV-1 replication are numbered. The HIV-1 replication cycle begins with viral entry into the target cell. Entry occurs upon fusion of the viral lipid envelope with the host cell plasma membrane, a process mediated by a non-covalent complex of viral (Env) glyco-proteins gp120 and gp41. First, gp120 binds the cellular receptor CD4, and then interacts with the CCR5 or CXCR4 coreceptor. Coreceptor binding by gp120 triggers a series of conformational changes in both gp120 and gp41 that lead to membrane fusion. Following fusion, the viral core, composed of a capsid (CA) shell containing the dimeric single-stranded RNA genome in complex with the reverse transcriptase (RT) and integrase (IN) enzymes, is deposited into the cell. The core uncoats, and RT copies the RNA genome into a double-stranded DNA copy. This viral DNA is then transported into the cell nucleus where it is integrated into the host cell genome by the IN enzyme. Subsequent transcription and translation lead to production of the viral components that assemble into new particles. The assembly process, which occurs at the plasma membrane, is directed by the Gag polyprotein precursor. To promote the budding and release of newly assembled virus particles, HIV-1 hijacks cellular endosomal sorting machinery (referred to as the “ESCRT” complexes) that normally functions to promote the budding of vesicles into late endosomes to form multivesicular bodies. Following release, the viral protease (PR) cleaves the Gag polyprotein precursor into individual Gag domains, thereby triggering a maturation process that is absolutely required for particle infectivity. Host factors that play a positive role in HIV-1 replication are indicated in green; factors that can restrict HIV-1 replication are in red. Adapted and reprinted with permission from Elsevier (1), © 2004.
Footnotes
-
↵1 Whereas a CC chemokine (or β-chemokine) contains two adjacent cysteines near its N terminus, a CXC chemokine (or α-chemokine) contains an amino acid between the two cysteines.
- © American Society for Pharmacology and Experimental Theraputics 2009
References

Catherine S. Adamson, PhD, is currently a postdoctoral fellow in Eric Freed’s laboratory within the HIV Resistance Program, National Cancer Institute (NCI) in Frederick, MD, USA. She received her doctoral degree from the University of Manchester, UK with Jayne Brookman. Before joining Eric Freed’s lab she performed postdoctoral research with Professor Ian Jones at both the Oxford Institute of Virology and the University of Reading, UK. Her research interests focus on retrovirus particle assembly, release, and maturation. Recently she has made significant contributions to development of the novel HIV-1 maturation inhibitor, bevirimat. E-mail cadamson{at}ncifcrf.gov; fax 301-846-6013.

Eric O. Freed, PhD, is currently Chief of the Virus-Cell Interaction Section of the HIV Drug Resistance Program, National Cancer Institute (NCI) in Frederick, MD. He received his doctoral degree from the University of Wisconsin, Madison with Rex Risser and performed postdoctoral research with Howard Temin in Madison and Malcolm Martin at the NIAID/NIH in Bethesda, MD. He received tenure in the NIAID in 2002 and in 2003 moved to the NCI. His research focuses on the molecular biology of HIV-1 replication. He has made a number of key discoveries in the field of HIV-1 assembly and release. E-mail efreed{at}nih.gov; fax 301-846-6777.

