Chemokine Signaling and the Management of Neuropathic Pain
Abstract
In the 1930s, presumably after the repeal of prohibition, laborers who came to work at a certain rubber factory found that a drink at the end of the working day did little to relax them. Flushing and nausea, vertigo, headache, and hypotension were the more obvious effects of the workers’ intake of alcohol. It turned out that exposure to disulfiram, a chemical used at the rubber factory, made the imbibing of alcohol aversive. By 1951, disulfiram was dispensed as a drug, named Antabuse, for the treatment of alcoholism. Disulfiram functions as an inhibitor of aldehyde dehydrogenase, and the accumulation of acetaldehyde in patients who receive disulfiram and then drink alcohol is the basis of the disulfiram–ethanol reaction. So why should disulfiram, clearly implicated in inhibiting ethanol metabolism, be effective in treating cocaine addiction? At first consideration, a number of psychological and social explanations come to mind: cocaine use is comorbid with alcohol abuse; the psychological deterrent created in treated alcoholics is transferred to the effects of cocaine intake. A series of clinical trials has established that disulfiram is specific for cocaine use per se; indeed, disulfiram inhibits dopamine β-hydrolase and thereby plays directly to the mechanisms of cocaine reward. But disulfiram is no cure for addiction; enzyme inhibition by disulfiram as it is understood cannot address the psychosocial and biological underpinnings of addictive behaviors. Nevertheless, clinical trial data and neuropharmacological insights from disulfiram research enable investigators to make specific hypotheses for best treating the complexity of addiction.
Introduction
The sensation of acute pain (nociception) is clearly of great importance for the survival of all complex organisms. However, in a large number of clinical situations patients can develop chronic pain syndromes in which the sensation of pain is no longer associated with normal and physiologically appropriate aversive stimuli. Such “pathological” pain can result from damage to the nervous system, the toxic effects of drugs, infection, and several other causes. Under these circumstances patients develop spontaneous sensations of pain and also experience pain in response to stimuli that are not normally perceived as noxious (i.e., allodynia), such as light touch or temperature variation. The chronic and intractable nature of these syndromes can greatly compromise the quality of life of affected individuals. Although drugs are available that afford a degree of relief to some individuals, they are not universally effective. Powerful pain-killing drugs, such as opiates, may produce some relief but can over time result in serious opioid-related problems such as tolerance and dependence. Indeed, many chronic pain syndromes remain difficult to manage pharmacologically.
It is widely believed that the development of chronic pain behavior involves fundamental changes in the properties of the neurons that signal pain, including the sensory neurons of the dorsal root and trigeminal ganglia as well as neurons higher up the neuraxis. It appears that the complement of neurotransmitters, receptors, and ion channels is altered in certain dorsal root ganglion (DRG) neurons, resulting in hyperexcitability and ectopic firing, which is perceived as a spontaneous sensation of pain (1). In addition, changes in synaptic transmission at the level of the dorsal horn of the spinal cord (interneurons), projection neurons in lamina I and V, and neurons higher up the neuraxis (descending controls) result in central synaptic sensitization to pain-related stimuli (2). A fuller understanding of the molecular and cellular mechanisms that underlie these changes is essential to the development of more effective treatments and, ideally, a means for permanent reversal of chronic pain states.
Because chronic pain is such a widespread health problem, it has been intensively investigated in both academic and industrial laboratories, leading to many new insights; however, these advances have yet to be integrated into a logical framework for improving the treatment of chronic pain. It is clear that the stimuli that initiate chronic pain syndromes initially provoke some aspect of the inflammatory or innate immune response (3). Inflammatory responses, having evolved to adapt to general noxious stimuli, can be considered along a continuum that starts with the activation of resident tissue macrophages together with the expression of inflammatory cytokines. Depending on the severity of the stimuli, these initial responses may subsequently lead to the influx of additional leukocytes into tissues. Typically, such inflammatory responses will counter the insult to the organism, and the organism will once again return to a state of homeostasis. However, if the insult upon the organism is not resolved, then the chronic nature of the inflammatory response will in itself often result in tissue dysfunction (4), one manifestation of which appears to be chronic pain. If chronic pain is viewed in this way, in terms of a continuum of inflammatory processes, we may find points that represent novel targets for therapeutic intervention.
Chemokines and Chronic Pain
The relationship between the initiation of the inflammatory response and the development of chronic pain is now well established (5). This relationship is clearly mediate by the activation of a number of cell types with immune competency, including: Schwann cells; satellite glia cells within the DRG; macrophages in close proximity to peripheral nerves; and astrocytes and microglia in the vicinity of primary afferent central projections. Presumably, interference with the activation of these cellular contexts would prevent or limit the development of chronic pain.
The cascade of inflammatory events associated with both non-neuronal and neuronal elements includes a number of cell signaling events that may help us identify molecular mechanisms leading to chronic pain behavior. For example, cellular proteins connected with stress responses, or molecular components derived from bacteria and viruses, activate Toll-like receptors that are expressed on the surface of resident macrophages or microglia. This activation initiates a complex cascade of intracellular signals, including the expression and inflammatory cytokines that are secreted by the cell (6). Secreted cytokines, such as tumor necrosis factor alpha (TNFα) and interleukin-1 beta (IL-1β), participate in numerous functions of a variety of cells, often including the synthesis of numerous downstream mediators that orchestrate cellular immune responses (7).
In many inflammatory responses, the requirement for increased leukocyte participation is predicated upon the expression of small secreted proteins known as chemokines. Chemokines attract leukocytes to the site of injury by activating specific G protein–coupled receptors on the leukocyte cell surface. The role of chemokines in directing the migration of leukocytes appears to have evolved from a more ancient function in regulating the migration of tissue stem cells. The chemokine-mediated retention of hematopoietic stem cells in the bone marrow is an example of this earlier function, but the large number of vertebrate chemokines are predominantly associated with leukocyte biology. Nevertheless, it has recently been observed that chemokines and their receptors are also expressed by multiple cell types in the nervous system, indicating potentially unappreciated roles for chemokine signaling in neuronal cell function. For example, stromal-derived factor 1 (SDF1, also known as CXCL12) and its receptor (i.e., CXCR4) regulate neural stem cell migration and development and thereby reflect the evolutionary origins of chemokine signaling (8). Indeed, other roles for chemokine signaling are played at the interface of inflammation and nervous system function, as is clearly demonstrated in pain states. In this regard, monocyte chemoattractant protein 1 (MCP1, also known as CCL2) is a chemokine that, along with its receptor (i.e., CCR2), appears to be of particular importance (9). One result that clearly demonstrates this importance is the phenotype of knockout mice deficient in the CCR2 receptor (10). These mice exhibit a reduction in the development of neuropathic pain that is nearly absolute in some injury paradigms (e.g., sciatic nerve ligature; see below). Additionally, the overexpression of MCP1, or administration of the chemokine to rodents, produces manifestations of pain hypersensitivity (10–13). The molecular and cellular bases for these results await elucidation. We shall now review different types of data that illustrate the potential roles of CCR2 signaling in chronic pain behavior.
MCP1/CCR2 Signaling Following Injury
Oh and colleagues observed the expression of numerous chemokine receptors in neonatal DRG neurons in culture and demonstrated that these cells were responsive to a wide variety of chemokine agonists (11). In particular, the HIV-1 coat protein gp120, which binds to specific chemokine receptors, not only excited cultured DRG neurons, but also elicited nociceptive behavior when administered in vivo (11). In this way, chemokines might have a direct or indirect role in mediating pain in the context of inflammation, a situation in which chemokine expression is strongly upregulated. Moreover, it was suggested that chemokines would primarily produce direct effects through the excitation of sensory neurons.
The implication of chemokine signaling in chronic nociception was especially strengthened by the demonstrated the necessity of CCR2 signaling for a variety of acute pain behaviors such as the mechanical allodynia associated with inflammatory adjuvants. Significantly, CCR2 knockout mice did not exhibit mechanical allodynia following spared nerve ligation and intraplanar administration of MCP1. More recent results have demonstrated that mice that overexpress MCP1 show enhanced pain sensitivity (12), and drugs that block CCR2 receptors can inhibit established pain hypersensitivity (14–16) (Figure 1).
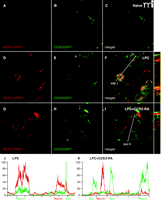
CCR2 signaling is activated in the DRG. Mice expressing MCP1 tagged with monomeric red fluorescent protein-1 (MCP1–mRFP1) were crossed with mice expressing CCR2 tagged with extra green fluorescent protein (CCR2–EGFP) mice. The resultant transgenic mice were subjected to lysophosphatidylcholine-(LPC)-induced demyelination of the sciatic nerve. In the DRG ipsilateral to the injury, expression of both MCP1 and CCR2 increased (D–F), whereas there was little expression of MCP1 or CCR2 under naive conditions (A–C). MCP1–mRFP1 mainly localized to neurons (large red arrow) and, to some extent, to satellite glia [small red arrow (D)]. CCR2–EGFP localized to neurons (large green arrow) and satellite glia [small green arrow (E)]. Most CCR2–EGFP-expressing neurons and satellite glia also contained MCP1–mRFP1 [yellow arrow (F)]. Injection of the CCR2–RA eliminated MCP1–mRFP1 in satellite glia [small green arrow (G–I)]. Also, after CCR2–RA treatment, MCP1–mRFP1- and CCR2–EGFP-expressing cells existed as separate populations [large green and red arrows(G–I)]. Cross-sectional images across the white lines are shown right to (F) and (I). (J, K) Intensities of mRFP1 and EGFP in shorter white lines in (F) and (I) are expressed in arbitrary units to compare relative signal intensities among different cells. (J) In the LPC group, most neurons that express CCR2–EGFP also contain MCP1–mRFP1. Also, the CCR2–EGFP signal in neurons is relatively weaker than the signal in satellite glia (J). (K) In the LPC plus CCR2–RA group, however, most CCR2–EGFP-expressing cells do not contain significant amount of MCP1–mRFP1 signal. In addition, the CCR2–EGFP signal in neurons is now as strong as the signal in satellite glia (K).
For a number of years, it has been believed that the primary role for MCP1 following peripheral nerve injury is to attract macrophages into the DRG and nerve as is seen in Wallerian degeneration. Indeed, it had been previously demonstrated that, in response to Wallerian degeneration, MCP1 was upregulated among nerve-associated Schwann cells and endoneurial fibroblasts (17). It was suggested that the elevated expression of MCP-1 in these cells played a central role in attracting macrophages into the injured nerve. Indeed, MCP1-expressing cells in injured sciatic nerve are reported absent in Wallerian degeneration–slow (WldS) mice (18), which are less susceptible to Wallerian degeneration. Moreover, WldS mice appear to lack the ability to develop thermal or mechanical allodynia following a chronic constriction injury (19, 20). In keeping with these considerations, Abbadie et al (10) observed a sustained influx of CCR2-expressing macrophages into the DRG and sciatic nerve in response to nerve injury; increased levels of mRNA for CCR2 were detected in the same locations. No expression of CCR2 was observed by neurons either in the DRG or spinal cord, although the authors detected immunoreactivity for CCR2 in microglia in the spinal cord of injured animals. Surprisingly, mRNA for CCR2 did not appear to be upregulated in the spinal cord of these animals, prompting the authors to propose a model in which MCP1 had two major functions. The first function is the attraction of macrophages into the DRG and nerve as observed in Wallerian degeneration; as a correlate to this function, it was proposed that infiltrating macrophages might secrete proalgesic factors. Secondly, as neuropathic pain is associated with the presence of activated microglia in the spinal cord, it was envisaged that injury-induced MCP1 synthesis might activate CCR2-expressing microglia, resulting in the release of additional proalgesic factors and accounting for cellular changes in the spinal cord that contribute to the development of chronic hypernociceptive states (21). In support of this latter proposal, the number of activated microglia and astrocytes appears to be reduced following nerve injury in the spinal cord of CCR2 knockout mice (10).
A somewhat different type of model was suggested by the finding that sensory neurons themselves might be the site of chemokine receptor activation in the generation of chronic pain, a suggestion that is in accord with the studies of Oh et al. (11). In two well-characterized neuropathic pain models, that is, partial ligation of the sciatic nerve and chronic compression of the DRG (CCD), MCP1 expression (mRNA and protein) was actually upregulated by the DRG neurons themselves (22, 23). These observations were consistent with other studies demonstrating that MCP1 could be upregulated in different types of neurons in response to injury (24). Tanaka and colleagues (22) considered that the targets of MCP1 released from neurons might include macrophages following release of the chemokine from peripheral nerve terminals. It was also suggested that MCP1 might act upon the cell bodies of DRG neurons by a process of chemokine release within the ganglia, or that MCP1 might activate elements in the spinal cord, such as CCR2-expressing neurons or microglia following transport and release of the chemokine by the central projections of affected primary afferent neurons. Using the CCD model of neuropathic pain, White and colleagues examined the possibility that DRG neurons might be the site of MCP1 action (23). These authors observed that CCR2 receptors were expressed by the cell bodies of DRG neurons of diverse sizes in the injured and closely juxtaposed ganglia, three to five days after injury. Similar to previous observations (22), the origins of the MCP1 in the CCD-injured sensory ganglia were small-to-medium–diameter sensory neurons. Importantly, MCP1 excited neurons of all sizes in the DRG from injured but not control animals (23), an observation that was consistent with the CCR2 expression data. These investigations were further corroborated by Qin et al. (25) demonstrated that addition of MCP1 to cultured DRG neurons elicited release of calcitonin gene related peptide (CGRP; a nociceptor neurotransmitter) from these cells, presumably as the result of increased neuronal excitation (see below).
In summary, these data illustrate the fact that there are numerous sites at which MCP1/CCR2 signaling might potentially elicit proalgesic effects, including the periphery, within the DRG, and in the spinal cord. A variety of investigations, using multiple pain models, have highlighted the key roles of neuronal and non-neuronal cells in MCP1/CCR2-mediated pain. Several theories to account for MCP1 signaling have come from these investigations, which should necessarily be seen as mutually exclusive.
Injury-induced Influences of MCP1/CCR2 Signaling in Pain
As discussed above, in response to injuries of various kinds, MCP1 expression can be upregulated by various cell types of the peripheral nervous system, including Schwann cells, resident macrophages, endoneurial fibroblasts, and primary afferent DRG neurons. In order to further understand the role of neuronal MCP1 expression in particular, Zhang and De Konick (26) examined MCP1 expression following sciatic nerve injury. In these experiments, MCP1 expression was upregulated by DRG neurons quite rapidly (<1 day). In addition to this expression within the sensory ganglia, MCP1 was reportedly transported by neurons to the spinal cord, into terminals that also expressed the nociceptor peptide neurotransmitter CGRP. The ability of peptidergic sensory neurons to upregulate MCP1 following injury has been established (13). Moreover, neuronal MCP1 can effectively be released in an activity-dependent manner into the spinal cord dorsal horn (13). The release of MCP1 from central afferent projections supports the idea that chemokine-dependent microglial activation is a critical component for the pathogenesis of neuropathic pain. A salient point necessary for this conclusion is that the spinal cord dorsal horn contains CCR2-expressing microglial cells, although the specific cellular localization of CCR2 expression is not entirely conclusive. Nevertheless, many investigators have generally accepted this conclusion, based on the localization data published in the study of Abbadie et al. (10).
Further data supporting a role for CCR2 signaling in chronic nociceptive behavior comes from Zhang and colleagues (27), who used chimeric mice to examine the relative roles of resident spinal microglia and infiltrating blood-born macrophages in the generation of chronic pain following injury to the sciatic nerve. As did several other investigators, Zhang et al. (27) demonstrated that chronic pain failed to develop in CCR2 knockout mice. Perhaps more importantly, intrathecal administration of MCP1, which increases microglial activation and pain behavior in wild-type animals, failed to do so in CCR2 knockout mice. In contrast, development of pain proceeded when CCR2 was selectively knocked out in either the central (spinal cord) or peripheral (blood/bone marrow) compartment. Zhang et al. also demonstrated that circulating macrophages entered the spinal cord in association with injury and differentiated to become microglia. Much of this process was thought to be dependent on CCR2 activation of macrophages both within and outside of the nervous system and did not occur when MCP1 was neutralized.
From these observations, one may conclude that an early event in the generation of chronic pain is the upregulation of MCP1 signaling between DRG neurons and CCR2-expressing microglia in the spinal cord (10, 13). Moreover, MCP1 expressed by DRG neurons is transported to the periphery as well as to the spinal cord and can be released from nerve terminals following nerve excitation (13). CCR2 activation of microglia is thought to trigger the release of proalgesic mediators and substances that subsequently produce astroglial activation contribute to the eventual development of chronic pain (12). Influx of CCR2-expressing circulating macrophages in response to centrally released MCP1 may also contribute to the pool of activated microglia (27). Hence, it may be concluded that in order to therapeutically manage pain development, both central CCR2-expressing cells (microglia) and peripherally circulating macrophages must be targeted. Such a model clearly explains several key observations, including the role of CCR2 signaling as demonstrated by knockout mice, neutralizing antibodies, and CCR2 antagonist drugs. The model further sets the context for MCP1 upregulation and release from primary afferents, the observed requirement for activated microglia in the cord (as well as influx of macrophages), and the observation that MCP1 administration produces hypernociception. Essential to this type of model is that MCP1 should activate CCR2-expressing microglia in the spinal cord.
On the other hand, not all observations are completely consistent with the above model. For example, Abbadie et al. (10) and McMahon and colleagues (13) failed to demonstrate mRNA for CCR2 in the spinal cord or its upregulation in association with their injury models. Similarly, transgenic mouse models of in vivo MCP1/CCR2 signaling could not demonstrate the expression of CCR2 receptors by microglia either under resting conditions or in the context of pain (Figure 2) (16, 28). In fact, only a single report has actually demonstrated the expression of CCR2 by microglia in the spinal cord, using immunohistochemistry, seven days after a spared nerve injury (10). This immunoreactivity, in light of the conflicting data discussed above, may possibly reflect an epitope closely related to but distinct from CCR2. In fact, the only CCR2 positive non-neuronal cells in the central nervous system have been identified as CD4+ T cells (29).
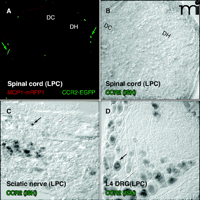
Peripheral nerve demyelination induces CCR2. CCR2–mRNA is induced in both the site of the nerve injury and in the sensory neurons of the dorsal root ganglion (DRG), but not in the associated spinal cord. (A) Neither the MCP1 monomeric red fluorescent protein-1 (MCP1–mRFP1) nor the CCR2 extra green fluorescent protein CCR2–EGFP in transgenic mice subjected to LPC-induced demyelination of the sciatic nerve exhibited expression of MCP1 or CCR2 at a detectable level. Leukocytes outside the spinal cord were clearly visible (green arrow). (B–D) CCR2 expression was also examined at the mRNA level by in situ hybridization. The spinal cord does not contain significant CCR2-expressing cellular components (B), whereas many cells in the sciatic nerve (C) and DRG (D) express CCR2 receptors in the LPC group. DH, Dorsal horn; DC, dorsal column.
Recently, two labs have detected the expression of CCR2 by neurons (but interestingly, not by microglia) in the dorsal horn, and these receptors would be accessible to MCP1 released by the terminals of sensory neurons in the dorsal horn of the spinal cord (28, 30). Using transgenic mice expressing CCR2-GFP, Gao and colleagues demonstrated, in agreement with previous studies, that CCR2 was upregulated by neurons in lamina within the deep dorsal horn following injury but was completely absent from the spinal cord of injury-naïve animals (28, 31). Gosselin and colleagues (30) provide the lone report that CCR2 is expressed in numerous neurons in the rat spinal cord under basal conditions. It is thus not yet certain whether low basal levels of CCR2 activation actually play a role in the genesis of chronic pain. Indeed, it has recently been reported that CCR2-expressing cells enter the central nervous system only when injury is paired with irradiation (as used in generating chimeras) (32). Thus, studies cited to support entry of CCR2-positive cells into the spinal cord [e.g., Zhang et al. (27)] may reflect experimental procedure, which greatly undermines conjecture that these cells enter the spinal cord in appreciable numbers in normal pain models.
Injury-Induced MCP1/CCR2 Signaling in the Periphery
If central activation of microglial cells does not explain the role of CCR2 signaling in hypernociception—can existing data provide an alternative explanation? One possibility is the importance of CCR2 signaling within the DRG itself. As discussed above, it is clear that MCP1 is robustly upregulated in numerous sensory neurons following nerve injury. Furthermore, it is apparent, under similar circumstances, that CCR2 receptors are also expressed by many closely juxtaposed DRG neurons (14–16, 23, 33). Thus, a model based on autocrine cell signaling or near-neighbor actions of MCP1 released from the DRG soma should also be considered. Several lines of evidence suggest that the direct effects of MCP1 on DRG neuronal excitability are of prime importance. First, it is clear that MCP1 and CCR2 are expressed by DRG neuronal cell bodies in close juxtaposition with one another following peripheral nerve injury. Second, MCP1 strongly excites CCR2-expressing DRG neurons, and such increased excitation is clearly an important feature of chronic pain behavior (23, 34). Third, the expression patterns for MCP1 and CCR2 in reporter mice are consistent with a role for either intersomal or intrasomal CCR2 signaling in the DRG. Using double-transgenic mice expressing distinctly labeled MCP-1 and CCR2, it can be observed, in several chronic pain models, that DRG neuronal cell bodies contain intracellular vesicles that are positive for MCP1 and CCR2 (25, 33), and the appearance of these vesicles can be inhibited by CCR2 antagonists (16). These observations are consistent with the view that activation of CCR2 receptors results in internalization of the receptor–ligand complex, in which case there may be constant release, binding, internalization, and recycling of MCP1 following its upregulation by DRG neurons. Given the fact that activation of CCR2 receptors expressed by these neurons is strongly excitatory, the constant release of MCP1 from the cell bodies of DRG neurons might then serve to spread an excitatory signal throughout receptive neurons in the ganglion (Figure 3).
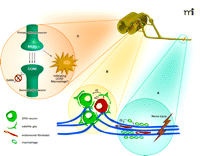
MCP1 as a chemokine in pain conditions. MCP1 may act at multiple sites and the precise nature of its involvement may differ in different pain conditions. (A) Schwann cells and/or endoneurial fibroblasts in injured axons upregulate MCP1, which then attracts macrophages into the nerve. Infiltrating macrophages may then secrete inflammatory molecules that sensitize the nerve. (B) Neurons in the DRG also upregulate both MCP1 and CCR2. The activation of CCR2 signaling in DRG neurons is excitatory and therefore pronociceptive. (C) DRG neurons may transport MCP1 to central nerve endings in the spinal cord where it is released. Once released in the spinal cord, MCP1 may activate secondary neurons and some CCR2+ leukocytes. Some neurons have been reported to express CCR2, and activation of CCR2 in these neurons may inhibit their response to GABAergic input (C).
The mechanism of MCP1-induced excitation is not entirely clear and will certainly depend on the class of DRG neurons involved; however, TRP, potassium, and sodium channels have been implicated (33, 34). Given the fact that upregulation of MCP1/ CCR2 by DRG neurons occurs some days following the initial injury (14–16, 23), it is also likely that this excitation is more important for the maintenance of chronic pain rather than its initiation. According to this view, the major site of action of MCP1 is within the DRG rather than in the spinal cord. Precise elucidation of the site of CCR2 activation in pain behavior is important, because the therapeutic inhibition of CCR2 signaling may depend on whether drugs can be developed to act centrally or peripherally.
Conclusions
MCP1 is a chemokine that represents novel therapeutic targets in chronic pain conditions, although its precise mechanism has not been definitively established. The potential effectiveness of its inhibition is evident from studies that have examined the administration of antibodies against MCP1, the use of CCR2 receptor blockers, and the production of CCR2 knockout mice. The identification of the site of action of MCP1 will be important in understanding the role of this chemokine in normal homeostasis as well as in the development of corresponding therapeutics. In addition to MCP1 and CCR2, other chemokines and receptors may also be regulated in the DRG to contribute to pain behavior under varying circumstances. Hence, an understanding of the regulation of chemokine signaling in the DRG and spinal cord in its entirety is of great importance in understanding the molecular changes underlying the behavior of sensory neurons in the context of chronic pain behaviors.

Acknowledgments
FAW was supported by the National Institutes of Health [Grants NS049136 and DA026040], and RJM was supported by the National Institutes of Health [Grants NS043095, DA013141, and MH040165].
- Copyright © 2009
References

Fletcher A. White, PhD, is an associate professor at the Stritch School of Medicine at Loyola University, Chicago, with a joint appointment in Cell Biology, Neurobiology and Anatomy, and Anesthesiology. His research group studies the sequence and nature of the neuroinflammatory events that govern neuron and non-neuronal communication in the sensory ganglia. Many of these events may be critical for the discovery of new mechanisms and targets for chronic pain treatment. Address correspondence to FAW. E-mail fwhite{at}lumc.edu

Richard J. Miller, PhD, is the Alfred Newton Richards Professor of Pharmacology at Northwestern Medical School in Chicago. He obtained his PhD at Cambridge University, UK, and subsequently worked for Burroughs-Wellcome & Company. He then joined the Pharmacology Department at the University of Chicago, , where he worked for twenty-five years before moving to Northwestern University in 2000. His laboratory works on receptors, signal transduction, and synaptic communication in the nervous system, as well as mechanisms of neurodegenerative disease.

Polina Feldman, BS, graduated from University of Wisconsin-Madison in 2007, where she studied biology and psychology. She is currently pursuing her doctoral degree under the direction of Fletcher A. White in the Neuroscience Graduate Program at Loyola University, Chicago. Her research focuses on the neuroinflammatory response in the peripheral nervous system following injury. Specifically, she is investigating the degree to which chemokines contribute to chronic pain syndromes.

