Emerging concepts from the recent literature
Specific Inhibition of a Clinically Relevant Drug-Resistant EGFR
Gefitinib and erlotinib are reversible kinase inhibitors that target the epidermal growth factor receptor (EGFR). These drugs are in advanced clinical development for non-small cell lung cancer. Acquired resistance to these drugs commonly occurs through mutation of the gatekeeper Thr790 residue of EGFR, and the T790M mutation is detected in fifty percent of resistant patients. In their recent Nature paper, Zhou et al. screened for irreversible kinase inhibitors that specifically target this EGFR mutation by examining selective growth inhibition of a gefitinib-resistant cell line. A new irreversible inhibitory activity was discovered with a greater than thirtyfold increase in potency, relative to previously described EGFR inhibitors, against the T790M mutant EGFR. At the same time, inhibition of the wild-type EGFR was reduced 100-fold. The new inhibitory activity was also effective in appropriate murine models of lung cancer. The structural basis for inhibitor selectivity against the EGF mutant receptor was revealed by co-crystallization; altered topology of the ATP binding cleft of the mutant enzyme allows formation of a Cys797 covalent bond. This paper is important because it demonstrates that selectivity can be achieved against a drug-resistant mutant EGFR and points to the potential pharmacological use of irreversible inhibitors. [Zhou, W., Ercan, D., Chen, L. et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 462, 1070–1074 (2009).] —M Nakashima, U Pittsburgh
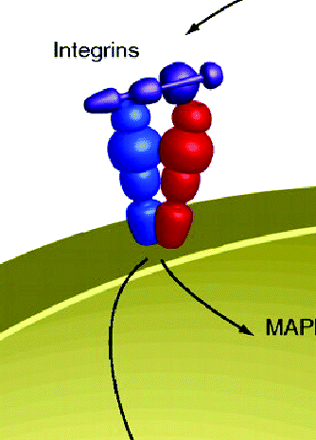
Gain-of-Function p53: Integrin Recycling and Metastasis
Drugs work best against gain-of-function targets. Thus, the loss of tumor suppressor protein, such as p53, which is inactivated in many human cancers through point mutation or enhanced degradation, is challenging to pharmacologists. Recently, Muller et al. report a mutant p53 gain of function in promoting cell mobility and invasion that is linked to enhanced integrin trafficking. The expression of mutated p53 proteins enhance invasion and migration of several cell lines and promote tumor metastasis in mice. The p53 mutant–driven migration and invasion depend on epidermal growth factor receptor (EGFR) signaling and the binding of β1 integrins to their ligands. Both ectopically expressed and endogenous mutant p53 are able to accelerate the recycling of integrin α5β1 and EGFR by promoting the association of α5β1 with the Rab-coupling protein (RCP). Furthermore, the migration and invasion effects may be a result of the ability of mutant p53 to inhibit the transcriptional function of TAp63. Although it is unclear how mutant p53 promotes integrin recycling, this study opens the possibility of designing therapies targeting gain-of-function mutant p53 by inhibiting integrin α5β1 and/or EGFR. [Muller, P.A.J., Caswell, P.T., Doyle, B. et al. Mutant p53 drives invasion by promoting integrin recycling. Cell 139, 1327–1341 (2009).] —F Zhang, U Pittsburgh
Living without Anxiety over the Risk of Benzodiazepine Dependence
Long-term use of benzodiazepines for the treatment of anxiety disorders often results in dependence. Alternative experimental drugs that could bind with differential affinity for benzodiazepine sites of the α1, α2, α3, and α5-subunits of GABAA receptors might result in nonsedating anxiolytics with lower abuse and dependence potentials. Ator and colleagues evaluated the abuse liability of two potential anxiolytics, TPA123 and TPA023, in baboons using self-administration tests. TPA123 showed weak partial agonist efficacy at α1, α2, α3, and α5-subunits, whereas TPA023 was devoid of efficacy at α1 and α5 subunits and showed weaker efficacy than TPA123 at α2 and α3 subunits. Dissimilar to TPA123, baboons did not self-inject TPA023, and after long-term high dose administration of TPA023, baboons showed only mild withdrawal symptoms. These findings support the concept that subunit-selective compounds can elicit different extents of abuse liability. Compounds with reduced efficacy at α2 and α3 subunits and no efficacy at the α1 subunit show therapeutic promise as anxiolytics with little to no abuse potential. [Ator, N.A., Atack, J.R., Hargreaves R.J., Burns, D., and Dawson, G.R. Reducing abuse liability of GABAA/benzodiazepine ligands via selective partial agonist efficacy at α1 and α2/α3 subtypes. J. Pharmacol. Exp. Therap. 332, 4–16 (2010).] —HN Brevig, U Michigan
Ethanol Use and Leaky Gut: Therapeutic Implications
The harmful effects of ethanol are primarily attributed to the formation of its highly reactive metabolite acetaldehyde. Pathophysiological levels of acetaldehyde in the gastrointestinal tract, seen in chronic alcoholics, may contribute to the formation of “leaky gut,” an increase in intestinal permeability that is associated with alcoholism. In a recent JPET article, Fisher et al. have investigated the effects of acetaldehyde on the paracellular permeability of hydrophilic markers in vitro and the oral bioavailability of several markers and drugs in the rat. The authors find that acetaldehyde significantly and reversibly increases permeability to mannitol and sucrose both in vitro and in vivo. Additionally, co-administration of ethanol and disulfiram, an inhibitor of acetaldehyde breakdown, significantly increases the in vivo bioavailability of the chemotherapeutic agent paclitaxel. These data highlight the importance of considering extrahepatic ethanol metabolism during drug treatment of chronic alcoholics, as the bioavailability of drugs with a narrow therapeutic window may be affected. [Fisher, S.J., Swaan, P.W., and Eddington, N.D. The ethanol metabolite acetaldehyde increases paracellular drug permeability in vitro and oral bioavailability in vivo. J. Pharmacol. Exp. Ther. 332, 326–333 (2010).] —NT Snider, U Michigan

- Copyright © 2010

