Personalized Medicine, Pharmacogenetics, and Clopidogrel: Unraveling Variability of Response
Persona est sui iuris et alteri incommunicabilis
—The Roman jurists
Among the currently marketed antiplatelet drugs, clopidogrel may display the most complex pharmacokinetics. Being a prodrug, the generation of clopidogrel’s active metabolite depends on the enzymatic balance between its bioactivation, mediated by the phase I enzyme cytochromes P 450 (CYP) and its bioinactivation, mediated by hepatic carboxylesterase (hCE)-1 (1, 2) (Figure 1). Two sequential CYP-mediated oxidations generate the active metabolite, whereas esterases inactivate 90% of the administered drug and nearly 50% of the intermediate thiolactone metabolite. Bioactivation is mediated by several CYPs, namely, 3A4, 2B6, 1A2, 2C9, and 2C19. The final, active metabolite of clopidogrel is an unstable, reactive, short-lived thiol derivative, which irreversibly inactivates the platelet P2Y12 receptor by forming disulfide bridges between cysteine residues, thus blocking ADP-dependent aggregation.
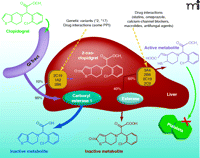
Of the different CYPs involved in activating clopidogrel, 3A4 is the most abundant in the human liver, and it metabolizes the greatest number of drugs, thus being the main player of the interactions between drugs, xenobiotics (food, chemicals), and hormones and the body’s responses to them (3). CYPs 2D6, 2B6, 2C19, 2C9 display clinically relevant genetic polymorphisms that affect their enzymatic activity. The activity of these isozymes may be also affected by xenobiotics, including drugs (3). Upon this highly complex pharmacokinetic background, the work of Shuldiner et al. has recently added to our understanding of individual variability in the responses of platelets and clinical outcomes to clopidogrel (4). One merit of this study was to consider a “genetically homogeneous” healthy population: the Amish. Another relevant aspect is the use of genome-wide association analysis, as compared to previous studies that considered a pre-defined set of CYP alleles. Patients at high cardiovascular risk treated with clopidogrel were studied in parallel. This study strengthens the (causal) association between the CYP2C19*2 allele, encoding for a loss-of-function enzyme, and a poor pharmacological response to clopidogrel, as assessed by a common, ex vivo method used to determine clopidogrel’s effectiveness at inhibiting platelet aggregation—light transmittance aggregometry (LTA) in response to adenosine diphosphate (ADP). Nevertheless, in the best possible, genetically uniform conditions, the 2C19*2 allele (in hetero- and homozygous status) accounts for only 12% of the variable response to clopidogrel, as measured by LTA. As in previous studies, non-carriers of the 2C19*2 allele were the large majority (70%), whereas 2C19*2 homozygotes, bearing the poorest response to clopidogrel, were just 2.1% of the population. Furthermore, there was a wide overlap of post-clopidogrel LTA values in non-carriers and 2C19*2 heterozygotes, with only 5–6% absolute difference in LTA values. Similar findings applied to patients. Moreover, more than half of the patients admitted, in follow-up to treatment, to prematurely halting their clopidogrel regimen, considerably reducing the sample size available to evaluate the clinical response as a function of the genotype. Six primary endpoint events occurred in the sixty-six non-carriers, whereas five events occurred among twenty-seven 2C19*2 carriers, with a hazard ratio (HR) of 3.4 and a wide 95% confidence interval (CI) (1.36–8.46). When LTA measurements were added to the regression analysis, the relation between 2C19*2 and cardiovascular outcomes was no longer significant (HR 1.58, 95% CI 0.68–3.66, p=.29).
This study raises several issues involving the application of pharmacogenomics and personalized medicine: 1) to understand further the wide variability in response to clopidogrel treatment in a population; 2) to predict the response of a single patient; 3) to determine clopidogrel’s clinical efficacy; 4) to evaluate the reliability and validity of the assays used to assess the genotype-phenotype relationship on antiplatelet drug treatment; and 5) to identify and minimize sources of response variability (i.e., such as obesity, circulating lipids and drug interactions).
Variability in response to drugs can be defined as “an effect of varying intensity occurring in different individuals at a specified concentration (dose) of a drug,” or as “a requirement of a range of concentrations (doses) in order to produce an effect of specified intensity in all of the patients” (5). Variable response is the final result of different components: 1) pharmacokinetics, including compliance, absorption, bioavailability, and biotransformation; 2) pharmacodynamics (i.e., molecular conformation and/or accessibility of the drug target, resistance, tolerance); 3) environment [such as interaction with xenobiotics, including drugs, habits (based on external cues)]; 4) physiological and pathological conditions (for example, age, kidney/liver function, underlying disease); and 5) genetic background. Indeed, genetic background may affect not only drug response but also disease onset, development, and interaction with the environment. How these determinants combine and affect the final drug response and the clinical outcome in the single patient is hard to predict and will be a significant challenge to research for years to come. As the drug armamentarium grows and the length of life increases, and because the environment is ever-changing, it is of utmost importance to identify sources of variability to optimize drug response and to minimize risks.
The multi-faceted and puzzling nature of factors affecting drug response may be decoded by the promise of “personalized medicine” (6). Personalized medicine has been defined as “the application of genomic and molecular data to target better the delivery of health care, facilitate the discovery and clinical testing of new products, and to help determine a person’s predisposition to a particular disease or condition” (7). This definition is shared by regulatory agencies such as the US Food and Drug Administration (FDA) (6). Personalized medicine is not a new approach in therapy; its beginnings can be traced back centuries and is based on common sense and empirical observations. Nonetheless, contemporary personalized medicine utilizes new technologies that are translated into drug development and clinical practice.
With regard to antiplatelet drugs, variability, pharmacogenomics, and personalized medicine are relatively new concepts. These concepts, however, have been associated for a much longer period of time with older drugs such as anticoagulants (warfarin), anticancers, and some antibiotics (anti-tubercolotics). The term pharmacogenetics was coined in 1959 by Vogel, who observed that isoniazide was differently metabolized, in an inheritable fashion, and this affected adverse reactions (see 8). Later, phase II biotransformation enzymes were identified as responsible for the “slow-metabolizer” phenotype (see 8).
Warfarin remains the leading cause of lethal drug reactions because of its complex pharmacokinetics and pharmacodynamics, high degree of intra-individual variability [such as, drug interactions or transient metabolic changes (e.g., fever, infections)] and inter-individual variability in dose requirements, and narrow therapeutic window (9). This profile, together with the absence of alternative drugs (10), have obliged the medical community to identify and standardize a test strictly dependent on drug concentrations and predictive of clinical benefit and bleeding risk. Such a test is the “prothrombin time,” standardized as the International Normalized Ratio (INR). The narrow therapeutic window of warfarin had forced physicians, as best they could, to tailor, the regimen to the individual patient—well before the characterization of warfarin pharmacogenetics—based on underlying disorders, metabolic status, concurrent drug or food intake, and age. Recently, the pharmacogenetics of key enzymes involved in warfarin pharmacokinetics (CYP2C9 alleles) and pharmacodynamics [vitamin K epoxide reductase complex 1, (VKORC1) alleles] has aided the precision of individualized treatment (9). Carriers of CYP2C9 loss-of-function alleles, being poor metabolizers, need lower doses, more time to achieve stable anticoagulation, and have an increased bleeding risk (9). Different VKORC1 polymorphisms are associated with either hypersensitivity or true resistance. The CYP2C9 and VKORC1 polymorphisms can, in total, explain up to 50–60% of the variable response to warfarin. On this basis, in 2007 the FDA added to the package insert that “lower initiation doses should be considered for patients with certain genetic variations in CYP2C9 and VKORC1 enzymes” (11). In addition, assays to identify the CYP2C9 and VKORC1 alleles have received FDA approval. Nevertheless, whether a pharmacogenetic-driven dosing of warfarin is safer and more efficacious than the traditional INR-based approach remains to be validated by ongoing trials (12). Therefore, routine genotyping is not currently recommended (10) because of a lack of proven evidence. Finally, the emerging oral anticoagulants under approval or advanced state of clinical development (13), are characterized by simpler, more predictable pharmacokinetics and pharmacodynamics, without need for monitoring. These new anticoagulants might, in fact, render irrelevant the necessity for routine, clinical use of warfarin pharmacogeneitcs.
In the case of clopidogrel, does our current pharmacogenomic knowledge predict successful drug dosages and regimens in different types of patients while avoiding the risk of bleeding? In investigating the relationship between pharmacogenomics and the pharmacodynamic phenotype, the reliability and validity of the methods used to assess phenotype (i.e., platelet response to drug) are crucial (just as with INR and warfarin). Shuldiner et al., by using ADP-LTA, noted the wide variability of LTA responses even at baseline before clopidogrel treatment. The high coefficient of variation of LTA with and without concomitant antiplatelet treatment, has been confirmed by several studies on healthy subjects or patients (14–19). The wide inter- and intra-individual variability associated with the LTA assay (independent of the agonists used), the lack of standardization and consequent poor reproducibility over time make the signal-to-noise ratio of this assay very low and the data often scarcely reproducible (14–19). Thus, it would have been interesting to investigate different assays in relation to the genotype and to the stability of the phenotype over time in the study of Shuldiner et al. If these authors had measured the LTA repeatedly over time, and compared additional, different methods [such as new point-of-care functional platelet assays, and the vasodilator-stimulated phosphoprotein (VASP) (see below)] would we have read the same genotype-phenotype-clinical outcome relationships? At variance with INR, there is no consensus on a reliable, reproducible, standardized and clinically-predictive assay (15) to assess platelet function for the development of pharmacogenomics and, more generally, of personalized medicine applied to antiplatelet drugs.
Several recent studies have examined the pharmacogenetics of clinical, pharmacological, or platelet function responses to clopidogrel (18–26) (Table 1); most of these have explored specific polymorphisms of the CYP3A, 1A, 2C, 2B sub-families. Few studies have investigated together pharmacokinetics, pharmacodynamics and clinical outcome. Available clinical studies are mainly retrospective, observational, and based on registries, with the exception of two studies (18, 19), which were part of large randomized clinical trials, although these trials were not designed to assess the relevance of pharmacogenetics as the primary endpoint. Not unexpectedly, the methods used to assess the pharmacodynamic response of platelets have differed among studies. The majority used ADP-induced LTA with diverse concentrations of ADP (from 5 to 20 μM), fewer studies used the VASP and/or the Verify-Now P2Y12 point-of-care assay. These latter methods are characterized by a lower assay-related variability and, possibly, higher sensitivity (16). Furthermore, the VASP is not a test that determines platelet function per se, but it reflects the phosphorylation state of the specific intra-platelet protein (VASP), a reaction that is influenced by P2Y12 inhibitors. Moreover, very few studies have used repeated measurements to validate the reproducibility of the “unresponsive” phenotype over time (17, 18), or measured plasma concentration of active metabolite (19–21). In spite of methodological differences, the 2C19 loss-of-function allele(s) (*2, *3, *4, *5 or *8, depending on the studies) was consistently associated with a lower degree of platelet inhibition as compared to inhibition in non-carriers. Across all different studies, heterozygous carriers (one loss-of-function and one normal allele), presented an intermediate phenotype, in terms of platelet inhibition or active metabolite concentrations, between homozygous (two loss-of-function alleles) and non-carriers, with values largely overlapping that of the non-carrier and homozygous groups (4, 18, 19, 21, 25). The difference between heterozygous and non-carriers were more pronounced with the VASP assay, as compared to ADP-LTA (20, 25, 26). Nevertheless, given the small percentage of homozygous (1.5–3% across different studies), a real estimate of the genetic “gradient” effect on platelet function (none, one, or two loss-of-function alleles) is difficult. Conflicting evidence on the degree of platelet inhibition are available on the CYP2C9 and 2B6 variants, while consistently negative results are reported for the 3A5/4 and 1A2 enzymes (see Table 1). Overall, these results appear consistent with a recent pharmacokinetic study, which explored the relative contribution of each CYP450 subfamily to the first and second reaction of clopidogrel bioactivation (1). The 2C19 participates in both enzymatic steps (Figure 1), accounting for approximately 45% of the total enzymatic activity in first reaction and for 21% in the second reaction, whereas the 3A4 enzyme is the main CYP participant in the second reaction (1).
Studies on CYP450 Variants, Clopidogrel Pharmacokinetics, Pharmacodynamics, and/or Clinical Outcome
Studies consistently reported no clinical impact for the 2C9, 3A4/5, 1A2 allele variants. In the study of Mega et al., the presence of at least one 2C19*2 loss-of-function allele was associated with worse outcome. In the study of Trenk et al (19), the influence of this same allele on death or myocardial infarction (MI), at one year post-treatment, disappeared when in the multivariate analysis included the ADP-LTA measurements. Simon et al., who performed the study with the largest number of patients per year, did not report any increased risk for a composite cardiovascular (CV) outcome (including death from any cause, nonfatal stroke, MI) in the carriers of one 2C19 loss-of-function allele. A significant clinical effect was observed only in patients bearing two such alleles (22). Therefore, two alleles appear to contribute to a worse clinical outcome as compared to non-carriers, while the estimate of the clinical risk of heterozygous carriers is more uncertain.
Given this evidence, which would be the best therapeutic strategy once the CYP450 genotype is known? Interestingly, in the TRITON-TIMI 38, the incidence of primary endpoints in the “non-carriers” of the 2C19 loss-of-fucntion alleles was similar to the incidence observed with prasugrel (8% vs 9.9%, respectively), indicating that non-carriers might similarly benefit from both drugs. On the other side of the spectrum, patients homozygous for the 2C19*2 allele might need up to 1.8 grams of clopidogrel to reach a platelet inhibition comparable to non-carriers (27). Notably, the majority (70%) of patients will still show a variable response to clopidogrel in spite of lack of 2C19 loss-of-function mutants. According to Shuldiner et al., only 10% of the variable response to clopidogrel can be attributed to the 2C19 genotype, while a similar percentage of variability depended on age, BMI, and lipid profile (4). Furthermore, as in the case of warfarin where pharmacogenomics might help preventing bleeding complications, data on 2C19 gain-of-function alleles (ultra-rapid metabolizers) were not associated with better cardiovascular protection or more bleedings (4, 21, 23) except in one recent report where the 2C19*17 gain-of-function allele was associated with a better clinical outcome (but the bleeding risk is unknown) (28).
The enzymatic activities of 2C19, 2C9, 2B6, are not only genetically determined but also drug modulated. Non-steroidal anti-inflammatory drugs and amiodarone are, for example, substrates of 2C9, nicotine is a substrate of 2B6, and clopidogrel itself is a moderate inhibitor of 2B6. Some proton pump inhibitors (PPI) inhibit 2C19, and the FDA has recently issued a warning on the interaction of PPIs and clopidogrel (29). As far as the 3A4 enzyme is concerned, its activity is predominantly modulated by drug (xenobiotic)-drug interactions, rather than by specific genetic variants (3). At least 50% of the marketed drugs are processed by 3A4, with poorly predictable kinetics. Therefore, drug interactions might induce a transient, modifiable, often unpredictable, “poor metabolizer” state.
Randomized studies assessing the safety and efficacy of genotype-based algorithms of treatment versus currently recommended treatment are lacking at the moment. Therefore, the pharmacogenomic/personalized era for clopidogrel is still in its infancy for routine clinical use. Supporting this point of view, a communication recently issued by the FDA, states that the agency “is aware of published reports that clopidogrel…is less effective in some patients than it is in others. Differences in effectiveness may be due to genetic differences in the way the body metabolizes clopidogrel” (30), without specific indications.
From a practical point of view, the CURRENT-OASIS 7 trial has recently shown the superiority of double vs standard dosing of clopidogrel (loading and maintenance) when given for a short interval to percutaneous coronary intervention (PCI)-candidate patients (31). The recently approved thienopyridine prasugrel, with a simplified pharmacokinetic profile (32), has shown less variable response, lessened influence by 2C19 genotype, and superiority vs clopidogrel in acute coronary syndromes (ACS) patients undergoing PCI, but at the price of more major (including fatal) bleedings (33). The non-thienopyridine, ticagrelor, which does not require bioactivation, was superior to clopidogrel in ACS, but non-CABG (coronary artery bypass graft) major bleedings were significantly increased (34). Thus, recently approved and future P2Y12 blockers appear increasingly less dependent of CYP450 genetic background. Whether a genotype-driven decision for a personalized clopidogrel regimen or for changing drug is clinically useful and economically convenient, remains to be confirmed by prospective studies. Ongoing randomized trials are testing whether a personalized dose based on residual platelet reactivity assayed at the beginning of clopidogrel treatment (35) can improve efficacy and safety, and results will be available within the next two years.
Some avoidable/modifiable factors [such as poor compliance, high body mass index (BMI), lipid profile], PPIs, lipophilic statins, and other drugs inhibiting the CYP450s are know to increase the variability in response to clopidogrel, affecting the salutary outcome for the patient. As also indicated by regulatory agencies, awareness of these elements and active intervention to correct modifiable factors in daily practice can narrow the variable response to clopidogrel in the individual patient, thus improving its efficacy. Pharmacogenetics is currently far from routine, but its development is crucial to understand the non-modifiable determinants of variable response to the “old” antiaggregants and is also relevant to guide the development of new drugs.
Acknowledgments
The Authors are grateful to Prof. Carlo Patrono for the critical reading of the manuscript and for his invaluable advices. Supported by the European Commission FP6 funding (LSHMCT-2004-005033).
- Copyright © 2010
References

Giovanna Petrucci, PhD, is Research Associate at the Department of Pharmacology of the Catholic University School of Medicine in Rome, Italy. In 1998 she received her BSc at the University “La Sapienza” of Rome, Italy. From 1999 to 2008 she worked as a Research Associate in the Laboratory and Clinical Unit for Immunohistochemistry at the Department of Pathology of the Catholic University of Rome. Her main research interest is in the study of the pathophysiology of platelets and megakaryocytes, and in particular in the expression and signaling of eicosanoid receptors and of cyclooxygenases during megakaryopoiesis and in circulating platelets. E-mail: giovanna.petrucci{at}rm.unicatt.it

Bianca Rocca, MD, PhD, is an Assistant Professor of Pharmacology at the Catholic University School of Medicine in Rome, Italy. She boarded in Hematology in 1995, was a Postdoctoral Fellow from 1995 until 1998 at the Center for Experimental Therapeutics of the University of Pennsylvania, in Philadelphia. From 1999 until 2004 she served as Clinical Fellow at the Clinical and Laboratory Unit for Disorders of Hemostasis at the Catholic University of Rome, Italy. From 2006 to 2008 she was Research Associate at the Center for Excellence on Aging of the University of Chieti, Italy. She is a member of the Working Group of Thrombosis and of the Working Group on Cardiovascular Pharmacology and Drug Therapy, of the European Society of Cardiology. Her main research interest is in the study of platelet activation and inhibition in atherothrombosis. E-mail b.rocca{at}tiscali.it; fax +39 06 305 0159.

