Pulse-Chase in the Light Microscope
To visualize specific proteins in living cells, cell biologists have fused the green fluorescent protein (GFP) or its analogs to the C termini of proteins and expressed these chimeras in culture and in embryos (1–,3). By using fluorescent proteins with different spectral properties (4–,6), researchers can follow exogenous proteins in real time and map their cellular locations and interactions.
These techniques have been complemented by the development of methods that permit the fluorescent labeling of recombinant proteins containing a tetracysteine sequence CCXXCC (where X is any amino acid). Proteins carrying this motif within an α-helix can be stained in vivo with the nonfluorescent, membrane-permeant biarsenical derivative of fluorescein, FlAsH-EDT2 [4',5'-bis(1,3,2-dithioarsolan-2-yl)fluorescein-(1,2-ethanedithiol)2]. FlAsH-EDT2 binds with high affinity (Kd ∼ 10 pM) to the tetracysteine motif, resulting in strong green fluorescence (7). Key advantages of this method are the smaller size of the tetracysteine tag, relative to the bulky ∼27-kDa GFP moiety, and the possibility of pulse-labeling the tagged proteins.
Gaietta et al. (8) have developed a further variation on the tetracysteine theme. In addition to the FlAsH-EDT2, they have synthesized ReAsH-EDT2, a biarsenical derivative of the fluorophore resorufin, which, when bound to the tetracysteine tag, emits a red fluorescence. Gaietta et al. (8) use these two reagents to label tetracysteine motif–tagged intracellular proteins, at spaced temporal intervals, permitting a visual pulse-chase in living cells. The method is especially useful because the ReAsH-EDT2 reagent is not only fluorescent, but also can be used, by photoconverting diaminobenzidine (DAB) to a highly insoluble reaction product, to visualize tagged proteins by electron microscopy (9).
Gaietta et al. (8) demonstrate the power of their novel method in a study of the gap junction protein connexin43 (Cx43), stably expressed as a tetracysteine-containing, recombinant protein in HeLa cells. The choice of Cx43 in gap junctions as a test specimen is particularly appropriate: Cx43 has a rapid half-life [1.5 hours (10)] and the protein accumulates at very high concentration in maculae at cell–cell appositions, greatly facilitating its visualization at the cell surface. Using GFP and its cyan (CFP) and yellow (YFP) color variants, Falk (11) showed that the products of different connexin-encoding genes can either co-mingle or segregate within individual plaques. In the stably transfected HeLa cells, the high expression of Cx43 favors the assembly of unusually large gap junctions, resulting in dramatically clear images as observed through the confocal light microscope. Gaietta et al. (8) first pulse-label tetracysteine-containing Cx43 in HeLa cells with FlAsH-EDT2. Following different chase times, newly synthesized, unlabeled Cx43 is then labeled with ReAsH-EDT2, such that the old Cx43 pool fluoresces green and the newly synthesized pool, red. The authors show by confocal microscopy that newly synthesized Cx43 is added to gap junctional maculae from the edges; the older Cx43 gradually disappears from a central domain within each macula (Figure 1⇓). These images are consistent with biochemical studies demonstrating that newly synthesized connexons (half-gap–junctional intercellular channels) are first inserted into non-junctional plasma membrane prior to their assembly into whole intercellular channels within the maculae (12). The images presented by Gaietta et al. provide strong evidence for lateral diffusion of the connexons in the plasma membrane in order to accrete at the edges of pre-existing gap junctions.
Gaietta et al. (8) further present data that exploit the ability to visualize ReAsH-EDT2 in the electron microscope. In one study, they first observe a gap junction by confocal microscopy. Then, subsequent to the photoconversion of DAB, the same junction is imaged using electron microscopy. The two imaging strategies were thus exhibited as equivalent. By utilizing the increased resolution inherent in electron microscopy on these overexpressing HeLa cells, they also detect newly synthesized Cx43 in small, putative post-Golgi transport vesicles [where Cx43 oligomerization is thought to occur (13)]. In addition, by reversing the staining order of the two reagents, senescent Cx43 in endocytic vesicles and secondary lysosomes can be clearly distinguished.
A powerful extension of the technique would be to use fluorescent resonance energy transfer (FRET) to monitor protein–protein interactions. In this approach, energy from one fluorescent molecule (the donor) is transferred by dipole–dipole interactions to a second molecule (the acceptor), provided that the two molecules are in a correct spatial orientation within 100 Å (14,15). This methodology has been used successfully by co-expressing GFP or its the cyan (CFP) or yellow (YFP) variants (16–,18). FRET could also be experimentally exploited by combining the CFP with FlAsH-EDT2 fluorophores. As a proof of concept, the tetracysteine-containing α-helix was fused directly to the C terminus of CFP and the resulting protein was modified with FlAsH-EDT2. The resultant complex displays FRET from CFP to FlAsH-EDT2 (7). The detailed time and spatial resolution achievable with the FlAsH-EDT2 labeling, combined with FRET, would potentially permit detection of transient protein–protein interactions and pinpoint these events within a specific intercellular compartment.
There are a few limitations to the method in its current form. First, although the authors engineered the tetracysteine motif into α-helical secondary structures, it is clear that the dyes can bind to endogenous cysteine-containing proteins. Cell types other than HeLa may show considerable background signals, limiting the method to only those proteins expressed at very high levels (19). It thus remains to be seen whether these probes can be effectively used in other cell types, or whether the method will be applicable to tetracysteine-tagged recombinant proteins expressed in transgenic animals. As seen in the study by Gaietta et al. (8), non-junctional connexons (12) are not visualized; only those connexons collected at high density in gap junctional maculae or in intracellular vesicles are visible. Thus, this method may not be applicable to study membrane proteins found at lower densities at the cell surface. Other proteins that do accumulate at high concentration in specific sites, such as in desmosomes, microtubules, actin and intermediate filaments, focal adhesions, chemical synapses, and tight junctions, will be readily amenable for similar studies, and the new approaches pioneered by Gaietta et al. (8) should provide some spectacular images in the near future.
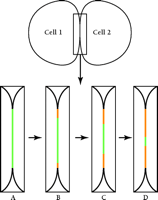
A diagram summarizing the visualization of pulse–chase of connexin 43 (Cx43) in gap junctions. Two cells are diagrammed; the boxed area of cell–cell interaction is enlarged in panels A–D. (A) Cells transfected with tetracysteine-tagged Cx43 are labeled with FlAsH-EDT2, which stains the gap junction fluorescent green. Following a wash, to remove excess FlAsH-EDT2, cells are treated with ReAsH-EDT2 to label all newly synthesized Cx43 fluorescent red. (B–D) Over time, newly synthesized Cx43 (red) replaces pre-existing Cx43 (green) by accretion at the edges of the gap junction with concomitant removal of connexons from the center.
- © American Society for Pharmacology and Experimental Theraputics 2002
References

Friso Postma, PhD, is a post-doctoral fellow in the Goodenough laboratory.

Daniel Goodenough, PhD, is the Takeda Professor of Cell Biology at the Harvard Medical School. Address all correspondence to DG: daniel_goodenough{at}hms.harvard.edu

