The Near-Death Experience of Delta Opioid Receptors Leads to New Drug Targets
The regulation of receptor expression at the cell surface is partially controlled by the endoplasmic reticulum (ER), which forms part of the secretory pathway. The ER is thought to process receptors constitutively and serve a passive role in signal transduction pathways, and as such, is not thought of as a target for direct therapeutic intervention. However, Petäjä-Repo and colleagues have challenged this assumption by demonstrating that cell-permeant pharmacological agents can interact directly with delta opioid receptors in the ER and rescue them from the degradative pathway.
The ER provides an environment that ensures the correct assembly and folding of proteins. Chaperones within the ER monitor the structure of newly synthesized proteins by recognizing properties, such as exposed hydrophobic sites, that distinguish misfolded proteins from their conformationally correct counterparts (1–,3). Proteins are sorted within the ER according to their folding and maturation status through a quality control process (3). Folded proteins move relatively rapidly from the ER—aided by the exposed amino-acid sequence motifs on protein surfaces that direct their sorting—into nascent COPII-containing vesicles that evaginate and bud off from the ER (1). Misfolded and unassembled proteins either aggregate or become degraded. The process of ER–associated protein degradation directs the movement of proteins from the ER to the cytosol where misfolded proteins are ubiquitinated and degraded by proteasomes (4).
One of the most common examples of a disease resulting from protein misfolding is cystic fibrosis. Mutations in the cystic fibrosis transmembrane conductance rectifier (CFTR) gene can lead to misfolding that compromises the trafficking of CFTR through the ER, and destines the mutant protein for proteasome-directed proteolysis (5). Chemical chaperones can rescue misfolded CFTRs retained in the ER (6), and pharmacological chaperones, such as vasopressin 2 (V2) antagonists can rescue mutant V2 receptors normally retained in the ER (2). Petäjä-Repo and colleagues now show that pharmacological chaperones can also rescue wild-type human delta opioid receptors (hδORs) (7).
The hδOR is very inefficiently processed, with approximately forty percent of newly synthesized receptor reaching the cell surface. Petäjä-Repo et al. had previously proposed that this receptor is unable to efficiently adopt the correct conformation, leading to its targeted degradation (8). Using pulse-chase labeling experiments, Petäjä-Repo et al. now demonstrate that cell-permeant opioid ligands interact with the intermediately folded precursors of the hδOR located on the surface of the ER (7). This treatment rescues the receptors from the degradative pathway and results in more receptors available for signaling at the cell surface. Both lipophilic opioid antagonists and agonists were able to rescue hδOR from the ER, suggesting that either ligand class could induce a more stable receptor conformation. Naltrexone treatment led to a twofold increase in the amount of mature (Mr ∼ 55-kDa) hδOR at the cell surface, while a concomitant decrease in the half-life of the core glycosylated precursor of hδOR (Mr ∼ 45-kDa) was observed at the ER. Additionally, naltrexone was able to rescue precursor hδOR that had been retained in the ER for up to four hours. It is provocative to note that ligands such as naltrexone function intracellularly at receptors located within the ER: cell-impermeant ligands, such as [Leu]enkephalin, had no effect on the trafficking of the mature receptor to the cell surface. Thus, the transport of receptors out of the ER using cell-permeant ligands may provide a new target for therapeutic interventions aimed at modulating receptor expression on the cell surface. Previously, it was not known why the chronic administration of opioid antagonists results in the greatly increased expression of the mu opioid receptor (9–,11). Petäjä-Repo and colleagues now hypothesize that increased surface expression results from the “pharmacological chaperone” properties of the lipophilic opioid antagonists.
The precise mechanism used by the ligands to rescue the precursor receptors is unknown; however, Petäjä-Repo and colleagues hypothesize that the cell-permeant ligands bind to the receptor precursors and stabilize conformations within the hydrophobic core of the protein. All cell-permeant ligands tested, which included opioid agonists and antagonists, were able to rescue hδOR, suggesting that active and inactive receptor conformations can be exported by the ER quality-control system.
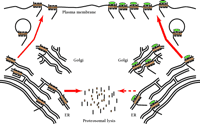
The fate of newly synthesized hδORs. On the left, receptors (represented by seven-transmembrane–spanning figures with yellow transmembrane domains) under endogenous conditions whereby only forty percent of the receptors reach the plasma membrane; the remaining sixty percent are retained in the ER, and are exported to the cytosol for proteasomal degradation. On the right, hδORs in the presence of a lipophilic hδOR ligand (represented by green-filled ovals) are processed through the ER more efficiently, so that fewer receptors are degraded and there is a concomitant increase in surface expression of receptor.
Receptor heterodimerization in the ER is important for the formation of functional γ-amino butyric acid subtype B (GABAB) receptors at the cell surface (12). On the other hand, a mutant CCR5 chemokine receptor that is retained in the ER can inhibit the exit of the wild-type CCR5 receptor (13). Opioid receptors exist as both homo- and heterodimers (14). Thus, perhaps pharmacological chaperones work by enhancing dimer formation within the ER. Further studies will be required to investigate this possibility.
The signaling capacity of the rescued receptors at the cell surface remains to be investigated. A number of other questions also arise: Are the ligand-bound receptors able to signal within the ER itself? Are there ligands that can destabilize receptors and effect their degradation from the ER rather than from the plasma membrane? Can pharmacological chaperones be designed that facilitate receptor maturation without interfering with binding of endogenous ligand once the receptor had arrived at the surface? It will also be important to establish whether pharmacological chaperones will work in neurons. The delta opioid receptors are located on dendrites and also in dense core vesicles. Is it possible that dense core vesicles are the proteasomal equivalent (i.e., the destination of the receptors that do not go to the surface) in neurons? Furthermore, it is still an open question whether all G protein–coupled receptors (of which opioid receptors constitute only a small number) can be rescued (and expressed at the cell surface) with appropriate pharmacological chaperones.
Pharmacological chaperones offer great advantages over chemical chaperones in terms of selectivity of action—that is, their inability to affect the folding and maturation of other proteins within the ER. For example, the treatment of pain may be improved by the co-administration of a lipophilic partial opioid agonist that operates as a pharmacological chaperone to increase the availability of receptors at the cell surface. Moreover, Petäjä-Repo et al. suggest that the quality-control system of the ER is amenable to regulation by pharmacological intervention, and in particular, the precise design of pharmacological chaperones may enable any receptor of interest to be targeted and retrieved from the ER (7).
- © American Society for Pharmacology and Experimental Theraputics 2002
References

Jennifer L. Whistler, PhD, (left) is a Prinicipal Investigator at the Ernest Gallo Clinic and Research Center; Adjunct Assistant Professor in the Department of Neurology, University of California, San Francisco; and is a Member of the Wheeler Center for the Neurobiology of Addiction. Address comments to JLW. E-mail: shooz2{at}itsa.ucsf.eduSelena E. Bartlett, PhD, (right) is a Senior Scientist at the Ernest Gallo Clinic and Research Center and is a member of the Whistler laboratory.

