I’ll Simply Die Without My Calcium: Ca2+ Signaling and Surviving Cellular Stress
Fungi are quite skilled at surviving all sorts of molecular insults, including our own pharmacological attempts to eliminate them. Thus, any pathway contributing to the hardiness of pathogenic fungi would be a reasonable and desirable target for antifungal drugs. Recently, just such a pathway has been described (1). Bonilla et al. have connected the known elements of calcium signaling pathways to the unfolded protein response (UPR), and in doing so, they have discovered a calcium signaling pathway involved in surviving multiple sources of cellular stress, fittingly labeled the calcium cell survival (CCS) pathway.
Calcium is essential for a number of processes occurring in the endoplasmic reticulum (ER). Depletion of calcium from the ER—for instance by using cell-permeant chelators—provokes a number of responses, including uptake of extracellular calcium and activation of calcium-dependent signaling pathways. Expanding upon observations made in mammalian cells (2), Bonilla et al. found that in yeast, depletion of calcium from the ER also induces another response: the UPR (1). The UPR is stimulated by the accumulation of unfolded or misfolded proteins in the ER and leads to the expression of genes that increase the protein-folding capacity of the ER (3). Surprisingly, in the absence of any chelators and in the presence of normal levels of extracellular calcium, genetic or pharmacological disruption of protein folding in the ER stimulates not only the UPR but also uptake of extracellular calcium and activation of calcium-dependent signaling pathways. Moreover, stress-induced calcium uptake and signaling are critical to the survival of cells suffering from the effects of unfolded proteins in the ER (1).
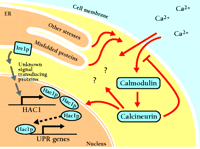
A schematic representation of the calcium cell survival (CCS) pathway. By unknown means, cellular stressors such as unfolded proteins activate calcium entry (black) and pathways such as the UPR (gray). Calcium enters through at least two mechanisms, one of which is the Cch1-Mid1 channel. Initial calcium entry initiates a chain of events (red), including the activation of calmodulin, which leads to calcineurin activation. Calcineurin modulates calcium entry, thus sustaining Hac1-dependent transcription and other unknown activities, and ensuring cell survival.
Bonilla et al. have identified four proteins required for the CCS pathway to function: calmodulin (CaM), the protein serine-threonine phosphatase calcineurin, and the plasma membrane calcium channel composed of Cch1 and Mid1. Another still-unidentified calcium uptake system is also involved, as some calcium accumulation occurs even in the absence of Cch1 and Mid1. Intriguingly, although CCS is activated in response to the presence of unfolded proteins, its activation does not depend on any known component of UPR signaling (1). Induction of the UPR begins with the action of the transmembrane kinase Ire1p (inositol-requiring 1) (4), leading to the production of transcription factor Hac1 protein (Hac1p, originally identified by its sequence similarity to transcription factors ATF and CREB) (5), which, in turn, increases transcription of the various genes that respond to unfolded proteins in the ER (3). Yeast lacking Hac1 or Ire1 accumulate calcium to nearly the same extent as wild-type cells.
Although activation of the CCS pathway does not depend on the components of the UPR, the CCS pathway might participate in the UPR. The initial stages of the UPR, including the production of Hac1p, do not require components of the CCS pathway. However, in a CaM-deficient yeast mutant that produces wild-type amounts of Hac1p, the Hac1p-dependent transcription of ERO1 was induced only transiently, suggesting that the CCS signaling bolsters Hac1p-mediated transcription (1). Thus, the CCS could be essential for sustaining what would otherwise be a temporary response. Nonetheless, the CCS must be doing more than simply enhancing the UPR, because the hypersensitivity of UPR-defective ire1 cells to tunicamycin (a toxin that blocks N-linked glycosylation of proteins in the ER) is further increased when calcineurin is inhibited (1).
The most important connection made by Bonilla et al. may be that the role of the CCS pathway extends beyond the response to unfolded-protein stress. It had previously been observed in the pathogenic yeast Candida albicans that the combination of cyclosporin (a calcineurin inhibitor) and the anti-fungal fluconazole was potently fungicidal, whereas fluconazole alone was only fungistatic (6). Earlier this year, Cruz et al. demonstrated that the synergistic action of the two drugs results from cyclosporin’s inhibition of calcineurin (7). The contribution of Bonilla et al. is their discovery that calcium uptake and signaling is the common requirement for surviving the inhibition of sterol production (by azoles, such as miconazole) and disruption of protein folding (by mutation or the glycosylation inhibitor tunicamycin). Yeast lacking the CCS pathway die at concentrations of miconazole or tunicamycin that only inhibit the growth of cells with a functional CCS pathway (1). Because miconazole treatment does not induce the UPR, we can conclude that the CCS functions more broadly in cell survival. That the CCS participates in cell survival shouldn’t be surprising, because the yeast genes involved in the CCS pathway are known to be essential in surviving pheromone-induced cell cycle arrest (8). The CCS pathway may be involved in the survival response to numerous other stresses as well.
Many details of the CCS pathway have yet to be identified, and chief among these is how the CCS promotes cell survival. One possibility is that the CCS acts to enhance long-term expression of a variety of stress-specific responses such as the UPR. Calcium signaling is known to induce transcription of a number of genes, and the ephemeral nature of Hac1p-dependent transcription in conditions of reduced amounts of CaM (the absence of the CCS?) certainly reinforces calcium’s critical role in transcription. But the mechanism of this CCS-mediated enhancement remains unclear, as the only known calcineurin-dependent transcription factor, Tcn1/Crz1, is dispensable for surviving conditions of unfolded protein stress. This Tcn1-independence seems particularly unusual in light of a recently published study finding almost all calcineurin-dependent gene expression in response to high Ca2+ or Na+ to be effected by Tcn1 (9).
A pathway so broadly involved in cell survival certainly appears to provide a useful approach to combating fungal infections. Although the synergy of calcineurin inhibitors and azoles is not new, perhaps further investigation will uncover even more powerful synergies for medical use. Regardless, experiments leading to a better understanding of the CCS pathway will certainly be welcome.
- © American Society for Pharmacology and Experimental Theraputics 2002
References

Stephen R. Cronin, PhD, is a Visiting Assistant Professor at the University of Nevada Reno. E-mail scronin{at}unr.edu; fax 775–784–1302

