Phospholipase C–γ1: A Phospholipase and Guanine Nucleotide Exchange Factor
The phospholipase C (PLC) family of cellular proteins participates in the intracellular signaling mechanisms utilized by diverse growth factors (GFs). A wide variety of growth factor receptor tyrosine kinases (RTKs) phosphorylate and activate PLC–γ 1; nonetheless, the biological role and necessity of this signal transduction element in mitogenesis has remained unclear. Indeed, whereas recent results point to a more important role of PLC–γ 1 in GF-induced mitogenesis than was suggested by initial studies (1) , the molecular mechanism by which PLC–γ 1 mediates GF-induced mitogenesis remains elusive.
Recently, Snyder and coworkers have reported that the Src homology 3 (SH3) domain of PLC–γ 1 acts as a guanine nucleotide exchange factor (GEF) for PIKE [phosphatidylinositol-3’ kinase (PI3K) enhancer] (2) . PIKE is a nucleus-localized small guanosine triphosphatase (GTPase) that stimulates PI3K activity in the nucleus and mediates the physiological activation by nerve growth factor (NGF) of nuclear PI3K activity (3) . This enzymatic activity accounts for the mitogenic properties of PLC–γ 1. Moreover, these results show that the mitogenic activity of PLC–γ 1 is entirely due to its SH3 domain rather than its phospholipase activity and its SH2 domains. These results challenge the prevailing wisdom and mechanism by which PLC–γ 1 was believed to mediate GF-induced mitogenesis and reintroduce the concept that SH3 domains can display enzymatic activity in addition to acting as a binding motif.
PLC–γ 1 contains two SH2, one SH3, one pleckstrin homology (PH), and two catalytic domains. All of these domains are important for the function of PLC–γ 1 and the regulation of GF-induced mitogenesis (Figure 1⇓). Through its SH2 domain, PLC–γ 1 can form complexes with PDGF receptors (PDGFRs) or EGF receptors (EGFRs) in vivo (4–8) . The formation of these protein complexes leads to phosphorylation of PLC–γ 1 on tyrosine residues and an increase in its enzymatic activity (9–11) . The SH3 domain of PLC–γ 1 has been implicated in targeting PLC–γ 1 to the cytoskeleton (12) . The SH3 domain of PLC–γ 1 can also bind the GTPase dynamin in vitro and in vivo (13, 14) and to proline-rich peptides derived from the Ras GEF Son-of-sevenless (Sos) in vitro (13) . However, the physiological relevance of these interactions has not been determined. The PH domain of PLC–γ 1 reportedly binds to phosphatidylinositol-3,4,5-trisphosphate (PIP3 ), suggesting that PLC–γ 1 can be targeted to the membrane in response to GF stimulation (15) .
Although the SH3 domain of PLC–γ 1 might be responsible for mediating GF-induced mitogenesis (16, 17) , what is most striking about the paper by Snyder and colleagues is the suggestion that PLC–γ 1 itself can directly act as a physiologically important GEF in the nucleus through its SH3 domain and this GEF activity accounts for the role of PLC–γ 1 in mediating NGF-stimulated cell mitogenesis.
Before the publication of the Snyder lab’s results (2) , the role of PLC–γ 1 in GF-induced mitogenesis had been supported by various studies. Most of these studies indicated that the mitogenic property of PLC–γ 1depends on its interaction with RTKs or on the activation of its enzymatic activity and subsequent signaling cascades (1, 18) . The intracellular injection of PLC–γ 1-specific antibodies blocks serum- and Ras-stimulated DNA synthesis, indicating that PLC–γ 1 is required for serum-induced mitogenesis and that PLC–γ 1 is downstream of Ras (19) . Another report has showed that inhibition of Ras activity—by the use of the dominant-negative Ras mutant N17 Ras—has no effect on GF-induced activation of PLC–γ 1 (20) . Recently, it has been shown that PLC–γ 1 is required for PDGF-induced DNA synthesis and c-Fos expression (21) , and that homozygous disruption of the PLC–γ 1 gene (Plcγ1 ) results in embryonic lethality at approximately embryonic day (E) 9.0 (22) . Additionally, PLC–γ 1 enzymatic activity is required for GF-induced mitogenesis, and the function of PLC–γ 1 in GF-induced mitogenesis may be to produce diacylglycerol (DAG), which subsequently activates most protein kinase C (PKC) isoforms (23–25) . Together, these results support a “phospholipase” model for PLC–γ 1 in GF-induced mitogenesis (Figure 2⇓). In this model, after RTKs are activated by GFs, the SH2 domains of PLC–γ 1interact with the phosphotyrosine sites on RTKs—this interaction results in the translocation of PLC–γ 1 to the plasma membrane. The PH domain of PLC–γ 1 might act to stabilize the binding of PLC–γ 1 to the plasma membrane. Binding of PLC–γ 1 to RTKs results in the phosphorylation of PLC–γ 1 and the further activation of its enzymatic activity (18) . Increased amounts of DAG are produced by activated PLC–γ 1 whereby PKC becomes activated and phosphorylates the protein serine-threonine kinase Raf (26–32) . Activated Raf stimulates mitogen-activated protein kinase (MAPK), eventually leading to the activation of transcriptional factors c-Jun and c-Fos and DNA synthesis.
Snyder and coworkers show that whereas the PLC–γ 1 SH2 domain and lipase activity are not required for NGF-induced cell mitogenesis, the PLC–γ 1 SH3 domain is necessary and sufficient to mediate NGF-induced cell mitogenesis (2, 3) . PLC–γ 1 translocates from cytoplasm to the nucleus in response to NGF stimulation. The nuclear PLC–γ 1 then binds to and activates PIKE through its SH3 domain, whereupon activated PIKE then stimulates nuclear PI3K activity. Thus, PLC–γ 1 mediates the NGF-dependent activation of nuclear PI3K activity. This enzymatic activity accounts for the mitogenic properties of PLC–γ 1. These results suggest a “GEF” model for PLC–γ 1 to mediate GF-induced mitogenesis (Figure 2⇓) and are consistent with the conclusions of an earlier study suggesting that the SH3 domain of PLC–γ 1 is responsible for its mitogenic properties (16) . Each of the above two proposed molecular mechanisms are supported by experimental results and may explain the role of PLC–γ 1 in GF induced cell mitogenesis, and these models need not be mutually exclusive. Therefore, whereas PLC–γ 1 may function in cell type specific manners to explain divergent results, it may be that this multidomain protein is in fact able to effect a mitogenic response by different pathways.
An important unanswered question that arises from the discoveries of Ye et al. (2) concerns how PLC–γ 1 translocates to the nucleus in response to NGF stimulation. Although truncation of one of the two PLC–γ 1 SH2 domains has no effect on the activation of PIKE and nuclear PI3K, it is not indicated whether this mutant protein becomes tyrosine phosphorylated or associated with the NGF receptor at the plasma membrane. Thus, it is not clear whether the direct interaction between NGF receptor and PLC–γ 1, the phosphorylation of PLC–γ 1, or both, is required for the translocation of PLC–γ 1 from cytosol to nucleus to activate PIKE. A recent report by Hung and coworkers shows that activated EGFR located at the plasma membrane relocalized to thenucleus within five minutes after its activation (33) . Based on this observation, it is tempting to speculate that the trafficking of the NGF receptor might provide a mechanism to translocate associated PLC–γ 1 to the nucleus. On the other hand, cytosolic PLC–γ 1 may be modified indirectly by NGF receptor may interact with other factors that effect its nuclear localization (Figure 2⇓). An intriguing possibility arises from the Snyder group’s earlier observation that PIKE interacts with Protein 4.1 (3) . Protein 4.1 associates with the adaptor protein pICln (34) , which interacts with the protein Skb1/PRMT5/JBP1 (35) , known to be a protein arginine methyl transferase (36) . Recently, a proteomics approach was used to show that pICln, Skb1, and protein 4.1 associate in a complex with spliceosomal Sm proteins in HEK293 cells (37) . Indeed, Friesen et al. and Meister et al. demonstrated that the methylation of Sm proteins—and, consequently, their protein–protein interactions—occur as a function of this pICln-scaffolded complex (38, 39) . Because the methylation of Sm proteins may regulate their own nuclear transport, it is tempting to speculate that the interaction between PIKE and protein 4.1 could indicate that methylation is somehow involved in the nuclear localization of PLC–γ 1 and the regulation of its interactions with PIKE (Figure 2⇓).
The finding that an SH3 domain functions as a physiological GEF extends the functional role of SH3 domains beyond that of being mere protein-binding regions. For example, an SH3 domain of Grb2 can bind to dynamin and stimulate its GTPase activity (13) . Because the interactions between SH3 domains and proline-rich motifs (and structures similar to proline-rich motifs) occurs between many proteins, it will be interesting to see whether the SH3 domain of other proteins also have enzymatic activity and whether the GEF activity of the PLC–γ 1 SH3 domain shows specificity for PIKE. Another intriguing finding by Snyder’s lab is that PIKE-binding activity by an SH3 domain does not necessarily equate with PIKE GEF activity. SH3 domains from Fyn and Grb2 associated efficiently with PIKE, but did not stimulate PIKE GTPase activity. Hence, whereas conventional polyproline binding activity is apparently required for GEF activity by the PLC–γ 1 SH3 domain, this module may contain additional GEF determinants required to stimulate nucleotide release with PIKE. Alternatively, the GEF activity of PLC–γ 1 SH3 may reflect a more stable association with PIKE than achieved by the other SH3 domains tested.
Clearly, the findings of Snyder and coworkers provide interesting new insight into the function and mechanism of action of PLC–γ 1, and the regulation of cell proliferation. Furthermore, they provide yet another important example of the modular nature and multifunctional properties of proteins involved in signal transduction.
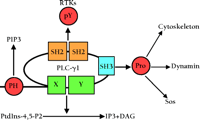
Organization of domains in PLC–γ1 and their interaction with other proteins and lipids. X and Y are catalytic domains of PLC–γ 1. PH, pleckstrin homology domain; PIP3 , phosphatidylinositol-3,4,5-trisphosphate; PIP2 , phosphatidylinositol-4,5-bisphosphate; Pro, proline-rich regions of various proteins; IP3 ; inositol trisphosphate; RTKs, growth factor receptor tyrosine kinases; pY, phosphotyrosine; DAG, diacylglycerol; SH2, Src homology 2 domain; SH3, Src homology 3 domain.
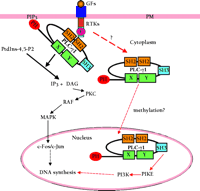
Two pathways depicting the molecular mechanisms by which PLC-γ1 mediates GF-induced cell mitogenesis. The red arrow indicates a guanosine nucleotide exchange factor (GEF) model [supported by (2)], and our speculation concerning methylation. The black arrow indicates a conventional phospholipase model that involves the PLC-γ1-dependent generation of DAG and IP3. GFs, growth factors; PM, plasma membrane; PI3K, phosphatidylinositol-3′ kinase; MAPK, mitogen-activated protein kinase.
- © American Society for Pharmacology and Experimental Theraputics 2002
References

Zhixiang Wang, PhD , is an Assistant Professor at the Department of Cell Biology, University of Alberta. Address correspondence to ZW at zwang{at}cellbnt.ualberta.ca

Michael F. Moran, PhD , is Chief Scientific Officer at MDS Proteomics, and an Associate Professor at the University of Toronto. Please address correspondence to MFM. Email mmoran{at}mdsp.com

