Targeting the FLICE Inhibitory Protein (FLIP) in Cancer Therapy
Cells of multicellular organisms have the inherent capacity to undergo inducible self-destruction by a highly organized process named programmed cell death, or apoptosis. This “suicidal” capability is utilized especially during development or upon injury or disturbance of tissue homeostasis. However, impairment of the apoptotic machinery of the cell or aberrant expression of anti-apoptotic proteins can contribute to the development of neoplasia and help tumor cells to escape from tumor surveillance by the immune system (1). A significant part of the benefit achieved by chemotherapy relies on the induction of apoptosis in tumor cells. Therefore, the development of resistance to apoptosis is of considerable clinical relevance (1). The central event in apoptosis is the proteolytic activation of cysteine aspartyl-specific proteases (caspases) that cleave defined cellular target proteins and thereby induce the characteristic morphological hallmarks of apoptosis. These effector caspases are triggered by initiator caspases that, in turn, become activated by associating with the signaling complex of death receptors or a cytosolic multiprotein complex called the apoptosome (2, 3).
Death receptors (e.g., Fas) represent a subgroup of the tumor necrosis factor (TNF) receptor (TNFR) superfamily and are structurally defined by an intracellular protein–protein interaction domain, called the death domain (DD), which is critically involved in apoptosis-induction by these receptors (4). Upon stimulation by their corresponding ligands, most death receptors interact with the DD-containing adapter protein FADD (Fas-associated death domain protein) through DD–DD association (Figure 1⇓). Death receptor-bound FADD in turn recruits the proenzyme form of caspase-8 (procaspase-8) thereby initiating the formation of the death-inducing signalling complex (DISC). Procaspase-8 is composed of an N-terminal regulatory prodomain consisting of two death effector domains, which are related to the DD, and a C-terminal caspase homology domain. In the DISC, several procaspase-8 molecules are located in close proximity, leading to their activation by dimerization (5,6). Subsequently, the active caspase-8 dimer is stabilized by a two-step autoproteolytic process (5,6). First, procaspase-8 (p55/53) is cleaved between the p20 and p10 subunits of the caspase homology domain generating a FADD-bound p43/41 intermediate, which is still associated with the p10 subunit (Figure 1⇓). Proteolytic cleavage of the p43/41 intermediate between the prodomain and the p20 subunit results in the release of mature, active caspase-8 (composed of a p20/p10 heteromer) into the cytoplasm. The FADD-bound prodomain can be finally replaced by another procaspase-8 molecule to start a new cycle of caspase-8 activation (4–6). Although caspase-8 activation can occur under nonapoptotic conditions, it typically leads to apoptosis by two different pathways. In “Type I” cells, caspase-8 directly activates effector caspases, especially caspase-3, to an extent sufficient to ensure apoptotic cell death (Figure 2⇓). In contrast, in “Type II” cells caspase-8-induced apoptosis relies on the stimulation of additional mitochondria-dependent amplification mechanisms (4).
Recruitment of mitochondria in apoptosis is initiated by caspase-8–mediated cleavage of the Bcl-2 family member BID. Upon cleavage, a fragment of BID translocates to the mitochondria and induces the release of apoptogenic proteins, including cytochrome c, second mitochondria-derived activator of caspase/direct IAP-binding protein (Smac/Diablo) and the serine protease HtrA2/Omi (7). Cytosolic cytochrome c triggers the assembly of the caspase-9 activating apoptosome, which in turn is able to activate caspase-3. Because caspase-3 can process and activate caspase-8 and caspase-9, a self-amplifying circuit of caspase activation is established (Figure 2⇓). Smac/Diablo and HtrA2/Omi block the caspase-inhibitory function of members of the inhibitor of apoptosis protein (IAP) family and thereby further facilitate the work of the effector caspases (7).
Each step in death receptor-mediated apoptosis is well regulated; however, the control of apoptosis imparted by the isoforms of the caspase-8-related FLICE-inhibitory protein (FLIP) is of particular interest. Although more than ten isoforms of FLIP mRNA have been described, only two of them, FLIPS and FLIPL, have been significantly studied at the protein level (6). FLIPL consists of two N-terminal death effector domains and a C-terminal caspase homology domain devoid of enzymatic activity, whereas FLIPS is only composed of the N-terminal death effector domains and a short C-terminal stretch of amino acids not found in FLIPL. Both proteins can be recruited to the DISC but they funtion differently: FLIPS prevents the initial cleavage step of caspase-8 activation between the p20 and the p10 subunit of the caspase homology domain; FLIPL inhibits the final cleavage between the prodomain and the p20 subunit of the p43/41 intermediate (Figure 2⇓).
There is growing evidence that FLIP can act as a tumor-progression factor (8, 9). For example, FLIP expression correlates with resistance against death receptor-induced apoptosis in a variety of B-cell lymphomas, and FLIP-transfected tumor cell lines develop more aggressive tumors in vivo (8, 9). Conversely, administering chemotherapeutic drugs to sensitize cells that are resistant to death receptor–induced apoptosis often correlates with decreased expression of FLIP (10). Additionally, FLIP is a target of the major anti-apoptotic pathways involved in carcinogenesis, namely the NF-κ B, Akt/PKB, and MAPK pathways (10). The particular relevance of FLIP for apoptosis-resistance has been pinpointed in recent reports showing that decreased expression of FLIP is sufficient to confer sensitivity against death receptor-induced apoptosis. In the human ovarian epithelial cancer cell line OV2008 transfection with a FLIP-specific antisense construct interferes with TNF-mediated, NF-κ B–dependent increases in FLIPS expression and confers sensitivity to the cytotoxic action of TNF, which is mediated by the death receptor TNF-R1 (11). Moreover, FLIP antisense oligonucleotides have been successfully used in three other studies to sensitize resistant prostate cancer cells (12), multiple myeloma cells (13), and chronic lymphocytic leukemia cells (14) to death receptor-induced apoptosis. A further report demonstrates that small interfering RNAs can be used for FLIP downregulation, too (15).
Inhibition of FLIP expression in tumor cells might be of particular importance for TNF-related apoptosis-inducing ligand (TRAIL)-based cancer therapies. TRAIL is the ligand of two death receptors, TRAIL-R1 and TRAIL-R2, and has attracted considerable attention in recent years as a potential anti-cancer reagent because of TRAIL’s ability to induce apoptosis preferentially in tumor cells (10). It is worth noting that TRAIL alone has little, if any, apoptotic effect and therefore requires cotreatment with conventional chemotherapeutic drugs that sensitize tumor cells, but unfortunately also some normal cells, for death receptor-induced apoptosis (10). Chemotherapy has often-pleiotropic effects, including the inhibition of anti-apoptotic pathways that typically regulate a whole battery of effector molecules. For example, the NF-κ B pathway induces, in a cell-type specific way, almost a dozen anti-apoptotic proteins including FLIP (16, 17). Therefore, it seems conceivable that chemotherapeutic drugs sensitize normal and cancer cells by targeting different cell-type specific effector proteins. Indeed, it has been recently shown that proteasome inhibitors sensitize primary keratinocytes to TRAIL-induced apoptosis by blocking the maturation and activity of caspase-3, most likely by interfering with the function of the inhibitory xIAP protein, an E3 ligase that drives the proteasomal degradation of caspase-3 and Smac/Diablo (18). However, proteasome inhibitors can also sensitize tumor cells for death receptor-induced apoptosis by blocking NF-κ B–dependent increased expression of FLIP (16, 17). This example illustrates that selective sensitizers of apoptosis might broaden the applicability of anti-cancer strategies related to death-receptor activation. Future studies must now show whether selective decreases in FLIP expression allow for differential sensitization of tumor cells and normal cells for death receptor-induced apoptosis.
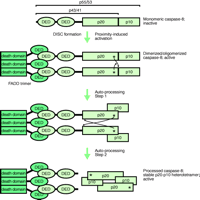
DISC-mediated activation of caspase-8.
Formation of the death-inducing signalling complex (DISC) initially activates unprocessed procaspase-8 by driving its dimerization. Subsequently, dimerized active procaspase-8 is autoproteolytically processed, attaining the mature heterotetrameric state of caspase-8 with DISC-independent activity. The thin arrows indicate proteolytic processing in trans, whereby the asterisks represent the active-site cysteine residues and the arrowheads point to the sites of proteolytic cleavage. However, upon their release by proteolysis—between the DEDs and the p20 subunits—the heterodimers orient themselves into a head-to-tail manner. FADD trimer bound to Fas (not shown). DED, death effector domain; FADD, Fas-associated death domain protein; p20 and p10 represent the subdomains of the caspase homology domain of caspase-8.
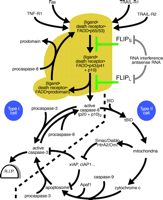
Apoptotic signaling pathways induced by death receptors and their regulation by FLIP.
In type I cells, death receptors (e.g., Fas) induce strong caspase-8 activation, which alone is sufficient to lead to robust processing of effector caspases such as caspase-3 and apoptosis induction (events shown left of the bold dashed line, golden brown region). In type II cells, only low amounts of caspase-8 are activated upon DISC formation. In addition, the action of effector caspases processed by caspase-8 is blocked by IAP proteins. Apoptosis induction in this type of cells is therefore dependent on a mitochondrial amplification loop, which triggers a second pathway leading to caspase-3 activation and furthermore interferes with the action of IAP proteins. IAP, inhibitor of apoptosis protein; BID, BH3-interacting domain death agonist; tBID, truncated BID (a caspase-derived cleavage product of BID); TRAIL, tumor necrosis factor–related apoptosis-inducing ligand; SMAC, second mitochondria-derived activator of caspase; Diablo, direct IAP binding protein with low pI; HtrA2, human serine protease with significant similarity to E. coli HtrA (HtrA2 is also called Omi); Apaf1, apoptotic protease activating factor 1.
- © American Society for Pharmacology and Experimental Theraputics 2003
References

Harald Wajant, PhD, is Professor and Head of the Department of Molecular Medicine at the Medical Polyclinic of the University of Wuerzburg. Address correspondence to HW. Email: harald.wajant{at}mail.uni-wuerzburg.de; fax +49 931 201 71070.

