COX-2 in Inflammation and Resolution
Abstract
Aspirin and the other NSAIDs have popularized the notion of inhibiting prostaglandins as a common anti-inflammatory strategy based on the erroneous premise that all eicosanoids are, within the context of inflammation, generally detrimental. However, our fascination with aspirin and the emergence of COX-2 has shown a more affable side to lipid mediators based on our increasing interest in the endogenous control of acute inflammation and in factors that mediate its resolution. Epilipoxins, for instance, are produced from aspirin’s acetylation of COX-2 and together with Resolvins and COX-2–derived prostaglandins of the D2 and J2 series represent an increasingly important family of immunoregulatory lipid mediators with strong implications for disease control and drug discovery.

Introduction: The Other Side of Inflammation
Inflammation, which occurs in response to an injurious stimulus, is a beneficial event that leads to removal of the offending factor and restoration of tissue structure and function. Thus, the hallmark of a successful inflammatory response is not only the clearance of injurious stimuli, but also the restoration of normal physiology. Stimulus clearance is mediated by a host of signals that drive the inflammatory response in a well-understood manner that involves the upregulation of cell adhesion molecules and chemokines, for instance, resulting in the accumulation of inflammatory leukocytes at the site of tissue injury. After the inflammatory stimulus has been neutralized, inflammation can abate. This abatement does not happen in a passive manner, as originally understood, through which the inflammatory response simply “fizzles out.” On the contrary, “resolution” of the inflammatory response is now appreciated to be an active event managed by an increasing number of recognized soluble mediators. These mediators engage mechanisms that culminate in the switching off of inflammation and return the stromal tissue that hosted the response to its prior physiology (1, 2). It may be anticipated, therefore, that failure of acute inflammation to resolve may predispose to auto-immunity, chronic inflammation, and excessive tissue damage, as shown previously (3).
Intriguingly, of the factors and signals inherent to the successful resolution of inflammation, the cyclooxygenase (COX) and lipoxygenase (LOX) pathways, through which arachidonic acid (AA) and other fatty acid precursors are metabolized, are essential (2, 4). This metabolic requirement is in stark contrast to the role of COX that has been established through the literature on nonsteroidal anti-inflammatory drugs (NSAIDs), according to which COX-derived AA metabolites are mostly pro-inflammatory. This article will examine the roles of diverse fatty acid metabolites and other signaling molecules in orchestrating the resolution of inflammation. New recognition of these roles will likely advocate for the development of drugs that are based on endogenous pro-resolution factors. At the very least, those in the business of developing anti-inflammatory drugs should be mindful of the existence of pro-resolution pathways and that unwittingly altering their normal functioning could come with unwanted side effects.
COX Metabolism and NSAIDs
Although AA may also be metabolized by cytochrome p450 systems, the focus of this review will be on the activities of COX and, to a lesser extent, LOX. There are three isoforms of COX, also known as prostaglandin G/H synthase. The first, COX-1, is constitutively expressed in most tissues and synthesizes prostaglandins (PGs) at low levels, and has thus been presumed to function primarily in the maintenance of physiological functions (5). COX-2, on the other hand, is highly inducible in response to pro-inflammatory stimuli, cytokines, and mitogens, and it has thus been generally conceptualized to function in the inflammatory release of PGs (6–10). Since the initial discovery of COX-2, however, these generalizations have been challenged. Phorbol esters, for example, support the differentiation of the THP-1 human monocytic leukemia cell line, in part by inducing COX-1 (7, 11), and stem cell factor and dexamethasone induce COX-1 expression in mouse bone marrow–derived mast cells (12). Furthermore, COX-2 is expressed constitutively in discrete populations of neurons throughout the forebrain, as well as in the cortex and hippocampus; its expression is similarly constitutive in the macula densa of the juxta-glomerular apparatus and adjacent epithelial cells of the cortical thick ascending limb of the kidney (6, 7). In endothelial cells, however, its expression is somewhat controversial; some workers maintain that COX-2 is expressed in the endothelium under resting conditions in response to shear stress, for instance, or under circumstances of focal inflammation (e.g., atherosclerosis), whereas others detect no COX-2 expression in the endothelial cells from various vascular trees (9, 13, 14). Finally, COX-3, a splice variant of COX-1, is inhibited by acetaminophen and is thus thought to mediate the antipyretic and analgesic effects of this drug (15); however, the protein expression and functional relevance of acetaminophen-sensitive COX-3 in humans has been questioned (16).
COX converts AA, which is released from the plasma membrane via the action of phospholipase A2, into PGG2 and then into PGH2 (Figure 1⇓). The latter, in turn, acts as a substrate for a series of downstream synthases responsible for the generation of PGs, thromboxane A2, and prostacyclin (collectively called prostanoids) (Figure 1⇓). Both COX-1 and -2 share similar structural properties, including a hydrophobic tunnel that allows AA access to the respective active sites. In COX-2, this hydrophobic tunnel has a side pocket, and the enzyme therefore manifests broader substrate recognition than does COX-1. This difference in substrate specificity will be important later when we discuss the ability of COX-2 to metabolize eicosapentaenoic acid and docosahexaenoic acid, either in the presence or absence of aspirin, to produce novel endogenous anti-inflammatory and pro-resolution lipid mediators. This structural distinction of the active site of COX-2 has lent itself to the discovery of compounds [i.e., the coxibs; e.g., rofecoxib (Vioxx, Merck)] that selectively inhibit COX-2 (17) and were thus developed and marketed as anti-inflammatory drugs. The coxibs, moreover, were reputed, by virtue of their limited gastric toxicity (18, 19), to have an advantage over traditional NSAIDs; it was initially believed that adverse gastric effects typically stem from the inhibition of COX-1. It is now generally believed that both COX-1 and COX-2 need to be inhibited simultaneously in order to elicit gastric ulceration, inasmuch as selective inhibition by coxibs only reduced ulceration by 50% compared to NSAIDs (18). In addition, the reported expression of COX-2 in the vascular endothelium, along with its roles in resolving acute inflammation and in healing gastric ulcers, has impugned the use of COX-2 inhibitors for treating chronic inflammation. Indeed, the association of adverse cardiovascular events associated with Vioxx consumption for chronic rheumatic pain has led to the voluntary withdrawal of Vioxx from the market. In the next few paragraphs, we will recount important evidence from the last ten years that shows COX-2 to be a versatile enzyme in the inflammatory response. This single enzyme possesses the ability, under certain conditions, to drive inflammation, and remarkably, the ability to resolve inflammation under other conditions.

The prostaglandin pathway. Synthesis of prostanoids by the COX enzymes following the catalytic release of arachidonic acid from membrane phospholipids by calcium-dependent phospholipase A2. PGH2 is metabolized as indicated by specific synthases. Alternatively, arachidonic acid can be metabolized by lipoxygenase or cytochrome P450 activities to leukotrienes, lipoxins, and hyroxyl/epoxyeicosatetraenoic fatty acids. Reproduced from (67).
NSAIDs and COX-2 Inhibitors
The inhibition of PG synthesis accounts, at least in part, for the anti-inflammatory properties of the NSAIDs that are widely used for the treatment of inflammatory joint diseases. The use of traditional NSAIDs, however, is also associated with renal and gastrointestinal toxicity arising from the inhibition of COX-1 (20). In this way, the discovery of COX-2 and the original belief that it was expressed only at sites of inflammation raised the possibility of selectively inhibiting this isoform and thereby exploiting the beneficial effects of NSAIDs without engaging their adverse effects. Consequent to the discovery of COX-2, the existing NSAIDs were classified according to their ability to inhibit either COX-1 or COX-2. Depending on the in vitro assay system used for screening, NSAIDs were generally shown to inhibit either COX-1 more selectively or both COX isoforms coincidentally (21).
COX inhibitors were then developed with up to 1000-fold selectivity for the inhibition of the “inducible” COX-2 isoform over the “constitutive” COX-1 isoform in various in vitro assay systems (22). Intriguingly, these COX-2 inhibitors showed little or no renal and gastric toxicity (23, 24). In terms of basic research, the development of selective COX-2 inhibitors resulted in the availability of various structurally diverse pharmacological tools, such as NS-398 (25), DUP-697 (26) and L-745,337 (23). The advent of effective COX-2 inhibitors, complemented by growing evidence that placed COX-2 at sites of inflammation and tissue injury, drove early studies that supported a pro-inflammatory role for this inducible enzyme (27, 28) and fueled speculation that its inhibition could me marshaled in the treatment of inflammatory diseases. In retrospect, however, the presumption that COX-2 was responsible solely for propagating the inflammatory response by synthesizing pro-inflammatory PGs appears specious.
A closer analysis reveals that the COX-2 inhibitors used in some of the early reports were administered at doses in excess of that required to inhibit COX-2. For instance, in one carrageenin air pouch study, the concentration of the highly selective COX-2 inhibitor, SC-58125, required to inhibit exudate formation was a 100-fold greater than that necessary to inhibit exudate PG levels (27). Significantly, this compound is 100-fold more potent as an inhibitor of COX-2 than of COX-1 in vitro; it is therefore possible that the SC-58125 doses used in this model inhibited COX-1 as well as COX-2. In addition to such pharmacological studies, the involvement of COX-1 in acute inflammatory models was similarly indicated in studies using knockout mice. AA induces edema in wild-type mice far more effectively than in COX-1–deficient mice (29), suggesting a role for COX-1 and not COX-2 in this model of inflammation. Additionally, COX-2–deficient and wild-type mice were found to have similar model responses in terms of ear swelling (induced by triphorbol myristate acetate) and paw swelling (in response to carrageenin) (30). Because mice devoid of COX-2 still express COX-1, it is conceivable that this constitutive isoform synthesizes sufficient bioactive PGs for inflammation to proceed. Collectively, these studies in COX-deficient mice indicate that both COX-1 and COX-2 can contribute to the inflammatory response.
Indeed, subsequent studies during the late 1990s began to suggest that in some tissues, most notably within the gastrointestinal system, COX-2 may be protective (31–33). In experimental models of colitis, for instance, the majority of PGs in the colonic mucosa are synthesized by COX-2 (34). Treatment with a selective COX-2 inhibitor (L-745,337) at doses that do not inhibit COX-1 depress mucosal PG synthesis and causes a marked exacerbation of colonic damage (35). Continuous treatment for up to one week results in perforation of the bowel wall and death. It was also shown that COX-2 mRNA and protein are strongly induced in murine models of gastrointestinal ulceration, concomitant with increased mucosal PG synthesis (36, 37). Treatment of the mice with the selective COX-2 inhibitor NS-398 reduces mucosal PG synthesis and significantly inhibits ulcer healing (38). These results have been confirmed by others who also show marked inhibition of gastric ulcer healing in rats by the COX-2 inhibitor L-745,337 at doses that selectively inhibit this inducible isoform (39). These studies thus question whether gastroprotective PGs are synthesized solely by COX-1 and suggest that COX-2 plays a significant role in protecting the gastrointestinal tract from injury. As mentioned earlier, it now seems that inhibition of both COX-1 and COX-2 in healthy gastrointestinal tissues is required to elicit ulcers.
Pro-resolution Properties of COX-2
The findings with NSAIDs and COX-deficient mice indicate that both COX-1 and COX-2 contribute to PG production at the site of inflammation and also that COX-2–derived PGs have roles in the resolution and healing phase as well as in the early stages of the inflammatory response. In the seminal paper on the phenotype of COX-2 knockouts, the mice developed suppurative peritonitis with multiple adhesions among the abdominal organs, consistent with chronic inflammation (40). In our laboratory, we have established that COX-2 is directly involved in regulating inflammatory resolution in a carrageenin-induced pleurisy model (41). Here we found two phases of COX-2 expression over the time course of this innate inflammatory response. The first peak (i.e., at about two hours) is associated with the onset of inflammation and is typified by polymorphonuclear leukocyte influx, PGE2 production, and COX activity (41); both the dual and the COX-2 selective inhibitors inhibit this early phase of the inflammatory response. However, forty-eight hours after irritant injection, we observed a second peak in COX-2 protein expression coincident with both the resolution of inflammation and the late influx of mononuclear cells. This second peak, moreover, manifests negligible PGE2 production but is associated with the clear presence of another PGH2 metabolite, namely, PGD2. Treatment with COX-2 selective inhibitors from between twenty-four and forty-eight hours post-injection of irritant appears to inhibit resolution and thus prolongs the inflammatory response; co-administration of PGD2 and cyclopentenone PGs, however, results in a normal course of resolution. We have also demonstrated that distinct phospholipase isoforms are differentially involved, with respect to the early versus late phases, in supplying substrate to COX (42).
PGD2 is derived from the metabolism of COX-derived PGH2 by hematopoietic PGD2 synthase (hPGD2S). In a more recent study, we sought to further define the role of COX-associated downstream synthases in inflammation by examining the role of hPGD2S in a model of lymphocyte-driven adaptive immunity elicited by methylated bovine serum albumin. Mice that are deficient in hPGD2S were found to display a more aggressive inflammatory response that fails to resolve, whereas mice overexpressing the enzyme display few signs of inflammation (43). Although hPGD2S-derived PGD2 and its metabolites, the cyclopentenone PGs, possess potent but diverse biological roles in host defense, the suppressive effects of hPGD2S on T-lymphocyte functioning in this model appears to be mediated by 15d-PGJ2 and its inhibition of NF-κB DNA binding (Figure 2⇓). Contrary to the emerging literature, however, there was little apparent role for PGD2, acting through either of its receptors [i.e., the D prostanoid (DP1) receptor and the chemoattractant receptor-homologous molecule expressed on Th2 cells (CRTH2)], in controlling lymphocyte functioning. These findings suggest an important role for both COX-2 and hPGD2S as checkpoint controllers in the progression from acute to resolving inflammation (Figure 2⇓). In ongoing work, we have found an equally important role for hPGD2S in the regulation of the onset phase of innate peritonitis as well as its resolution through the regulation of macrophage clearance to the draining lymphatics and regulatory lymphocyte functioning (R Rajakariar and DW Gilroy). This supports and complements the original findings with COX 2 in resolution of pleuritis as discussed above. Whether the absence of hPGD2S predisposes to chronic inflammation or auto-immunity has yet to be determined. Nonetheless, the clear lack of inflammation in animals that overexpress hPGD2S and the aggressive nature of the response in animals deficient in the enzyme further reinforce the critical role this downstream PGH2-metabolizing enzyme plays in the etiology of T-lymphocyte driven immune responses. Balanced against its pro-resolution role, PGD2 has been implicated in allergic inflammation as evidenced by its role in the pathogenesis of asthma (44). hPGD2S is abundant in mast cells (45) and dendritic cells (46), and its levels rise in the broncho-alveolar lavage following allergen challenge of asthmatic subjects (47), suggesting that the role of PGs in disease is highly context-dependent.
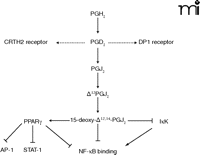
Actions of PGD2and 15-deoxy-Δ12,14-PGJ2. PGD2 acts via the DP1 receptor, which acts via adenylyl cyclase, and chemoattractant receptor-homologous molecule expressed on T helper 2 cells (CRTH2). DP1 receptor is expressed in mast cells and eosinophils. CRTH2 is expressed on a small population of CD4 and CD8 T lymphocytes and in peripheral blood eosinophils and basophils. See text for details. AP-1, a family of transcription factors; PPARγ, peroxisome proliferator-activated receptor γ; IκK, IκB kinase; STAT-1, signal transducer and activator of transcription 1.
Aspirin and COX-2 in Inflammation
As with the COX arm of the AA pathway, the LOX pathway is not wholly pro-inflammatory, with certain PG metabolites possessing either pro- or anti-inflammatory properties, depending on the given disease and organ. Leukotriene B4 (LTB4), for instance, is pro-inflammatory by virtue of its chemoattractant properties for polymorphonuclear leukocytes (PMN) (48); however, LOX-derived AA products may be further metabolized, by any of three pathways (Figure 3⇓), to generate distinctly anti-inflammatory and pro-resolving eicosanoids called lipoxins. Briefly, the first pathway, within eosinophils, monocytes and epithelial cells, involves the stereospecific insertion of O2 at carbon C-15 of AA. Following product release from these cells and entry into either PMN cells or monocytes, a 5,6-epoxytetraene is generated by 5-LOX, which is then hydrolyzed within these recipient cells by either LxA4 hydrolase or LxB4 hydrolase to produce bioactive LxA4 and LxB4, respectively (49). The second route of Lx biosynthesis results from the 5-LOX–catalyzed generation of LTA4 within leukocytes, its release and subsequent uptake by platelets, and metabolism by 12-LOX to yield LxA4 and LxB4 (50, 51). The third pathway entails one of the most exciting aspects of the mechanism of action of aspirin; by acetylating the active site of COX-2 in endothelial or epithelial cells, aspirin is unique among NSAIDs in triggering Lx synthesis to produce the so-called aspirin-triggered epilipoxins (52, 53). This third pathway results not in the inhibition of COX-2 enzyme activity, as might be expected, but in the conversion of AA into 15R-hydroxyeicosatetraenoic acid (15R-HETE) (54), which is rapidly metabolized by leukocyte 5-LOX, in a trans-cellular manner, to produce 15-epi-LxA4 or -LxB4.

Pathways leading to the production of lipoxins and epilipoxins. I). 15-lipoxygenase produces 15(s)hydroperoxyeicosatetraenoic acid [15S-H(p)ETE], which serves as a substrate for polymorphonuclear leukocyte (PMN) 5-lipoxygenase [5-LOX] to generate 5,6-epoxytetraene. The latter is unstable and is converted to lipoxin A4 and B4 by lipoxin (Lx) hydrolases. II). 5-LOX in PMNs metabloizes arachidonic acid to leukotriene A4 which is taken up by platelets in a transcellular manner and converted by platelet 12-LOX to LxA4. III). Acetylation of the active site of COX-2 by aspirin results in the conversion of arachidonic acid to 15(R)-hydroxyeicosatetraenoic [15(R)-HETE] which when released by endothelial and epithelial cells may be transformed by leukocyte 5-LOX to generate 15-epi-LxA4 or 15-epi-LxB4.
These three major pathways can operate independently or simultaneously within the vasculature. Neutrophils primed by granulocyte-macrophage colony-stimulating factor (GM-CSF) and recruited to the inflammatory site, for example, can interact with platelets. After platelets adhere to the neutrophil surface, active PMN cells generate LTA4, which is released and metabolized by platelet 12-LOX to generate lipoxins (51). Within the vasculature, the aspirin-triggered Lx pathway can also be initiated when activated endothelial cells interact with adherent neutrophils to generate 15-epilipoxins. Leukocytes interacting with epithelial cell surfaces, as in the case of respiratory, renal and gastrointestinal inflammation, can also generate lipoxins through bi-directional routes: 15S-HETE and 15R-HETE, released by epithelial cells, can be converted into lipoxins by neutrophils; neutrophil-released LTA4, on the other hand, is converted by 15-LOX in epithelial cells, in particular tracheal epithelial cells, also to generate lipoxins.
The lipoxins act via intracellular Ah (aryl hydrocarbon) receptor (55) or a G protein–coupled receptor termed ALX-R, or N-formyl-peptide receptor 1 (fprl1), expressed by neutrophils, monocytes, epithelial cells, and activated T cells as well as by enterocytes, fibroblasts, and the intracellular aryl-hydrocarbon receptor in dendritic cells (56, 57). Lipoxins are anti-inflammatory in their ability to inhibit neutrophil chemotaxis and diapedesis through post-capillary venules and therefore entry into inflamed tissues in animal models (58). In contrast, LXA4 also stimulates chemotaxis and adherence of monocytes to sites of inflammation (59), an essential mechanism in wound healing and clearance of leukocytes from inflamed sites. Attracted monocytes differentiate into macrophages capable of phagocytosing apoptotic neutrophils in a non-phlogistic manner (60), particularly during the resolution phase of inflammation, during which lipoxins promote monocyte chemotaxis and adherence without inducing neutrophil degranulation (61). 15-epilipoxins formed subsequent to aspirin administration [i.e., aspirin-triggered lipoxins (ATL)] share these properties in addition to other downstream beneficial effects that include increased vasorelaxation, prostacyclin synthesis, and induction of nitric oxide synthesis in endothelial cells (62). The benefits of lipoxins are potentially enormous and need to be exploited. The synthesis of stable analogs will be essential to elucidating the mechanisms of lipoxin action, and further understanding of the intrinsic mechanisms that underlie the ATL pathway is clearly needed.
Aspirin, COX-2, and Other Pro-Resolving Pathways
Resolvins and docosatrienes are fatty acid metabolites of the COX/LOX pathways, in which the omega-3 fatty acids docosahexaenoic acid and eicosapentaneoic acid, rather than AA, are the substrates (2, 61, 63). Resolvins and docosatrienes were identified in inflammatory exudates during the resolving phase of acute inflammation and shown to be potent modulators, at nanogram doses, of inflammatory processes. Specifically, they potently inhibit PMN transen-dothelial migration and microglial cytokine expression and also ameliorate experimental models of dermal inflammation and leukocyte accumulation in peritonitis (63 ,64). The studies on resolvin metabolism are uncovering surprising new avenues in anti-inflammation research, putting fatty acid metabolites right at the forefront of potential drug therapy. These studies are also challenging existing dogma by indicating that not all eicosanoids are detrimental to inflammation. Many therapeutic contexts are likely to benefit from this more balanced appreciation of the various roles that eicosanoids play in homeostasis and pathophysiology. To add fuel to this notion, a recent and very surprising paper has shown that eicosanoids of the lipoxin family are orally active in models of acute inflammation (65).
Conclusions and Implications for Drug Discovery
It is becoming clear that inflammation has a number of tactically placed checkpoints that limit the magnitude and duration of the response. Defects in these endogenous anti-inflammatory pathways will arguably predispose the host to chronic inflammatory diseases. The COX and LOX pathways have both protective and pro-inflammatory roles in inflammation, with the precise function of each enzyme being dependant on the inflammatory stimulus, phase of the acute inflammatory response, and other molecules that are present to tip the balance in favor of resolving or non-resolving inflammation. Our new appreciation for inflammation-related checkpoints raises a number of important matters pertaining to anti-inflammatory drug development, and we need to be mindful that during inflammation there may be multiple endogenous pathways attempting to limit or switch off the ongoing response. These pathways must not be inadvertently altered by “anti-inflammatory” drugs. The alternative benefits from this protective biological system is the realization that factors expressed during resolution could be mimicked and used to push ongoing inflammation down a pro-resolution pathway. Such an approach would entail the development of therapeutics that would exert multiple effects at various phases of the inflammatory response—for example, to limit leukocyte trafficking, hasten cell clearance, and help restore homeostasis to inflamed stromal tissue. It is provocative to think that existing anti-inflammatory agents, such as steroids whose development was based on an entirely different strategy, may exert potent pro-resolution effects arising from their pro-apoptosis (e.g., in the activation of eosinophils) and pro-phagocytic properties (66). The potential side effects of developing “pro-resolvers” might arise from the premature resolution of inflammatory responses before they have the chance to deal adequately with the insulting (i.e., “pro-inflammatory”) agent. In summary, all inflammatory responses are not the same, and pan-inflammatory inhibitors would not necessarily be the most effective approach to treating inflammation-driven diseases. We need to foster a greater understanding of the nature of the processes that drive each individual inflammatory response and tailor treatments accordingly. To achieve this goal, we must further elucidate the resolution of inflammation and identify possible approaches to promote this process in conjunction perhaps with anti-inflammatory therapies.
- © American Society for Pharmacology and Experimental Theraputics 2006
References

Magdi Yaqoob, PhD, is Professor of Nephrology at the Royal London Hospital and the head of Experimental Medicine, Nephrology and Critical care in the William Harvey Research Institute, London. His research interests include mediators of ischemia reperfusion injury, mechanisms of high glucose–induced proximal tubular injury, and mediators in the resolution of peritonitis.

Derek Gilroy, PhD, obtained his degree from the William Harvey Research Institute, University of London. His doctoral research addressed the role of inducible cyclooxygenase in inflammation, in collaboration with the late Professors Derek Willoughby and Sir John Vane. He pursued postdoctoral training with Dr. Kenneth Wu, jointly at the University of Houston Texas and at Academia Sinica, Taipei, Tawian. Now at the William Harvey Research Institute, he also holds an appointment as New Blood Lecturer, funded as a Wellcome Trust Career Development Fellow at the department of Medicine, Rayne Institute, University College London. His research interests focus on the endogenous factors that control the resolution of acute innate and adaptive immunity, as well as aspirin-triggered nitric oxide and the regulation of cell trafficking. Address correspondence to DWG. E-mail d.gilroy{at}ucl.ac.uk; fax +020-7679-6351.
Ravindra Rajakariar, PhD, (not shown) is a clinical research fellow in the department of experimental medicine, nephrology and critical care in the William Harvey Research Institute, London. His research has involved the role of PGD2 and cyclopentenone PGJ2 in the resolution of peritonitis in an experimental murine model and in end stage renal failure in patients on peritoneal dialysis.

