Tumor-associated, Estrogen Receptor-related Antigen EBAG9: Linking Intracellular Vesicle Trafficking, Immune Homeostasis, and Malignancy
- 1 Unit on Molecular Hormone Action, Program in Reproductive and Adult Endocrinology, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD 20892
- 2 First Department of Pediatrics, Athens University Medical School, Athens 11527, Greece
Recently, in an intriguing study, Rüder et al. reported that an estrogen receptor-related, ubiquitous intra- and extracellular protein produced at increased amounts by human cancer cells, acted as a novel inhibitor of the adaptive immune response, potentially influencing the growth and spread of malignancies (1, 2). Cytotoxic T-lymphocytes (CTLs) and natural killer cells (NKs) of, respectively, the adaptive and innate branches of the mammalian immune system, are crucial for immune surveillance and homeostasis. These cell types recognize and kill “non-self” cell populations, including tumor cells, cells infected by viruses or other pathogens, and those damaged by irradiation, toxic substances, etc. (3–5). CTLs recognize non-self cells via interaction of their specific T-cell receptors (TCRs) with foreign antigens co-expressed with the major histocompatibility (MHC) class I molecules on the surface of target cells. NKs, on the other hand, recognize non-self cells based on the absence of MHC class I expression on these cells, through receptors expressed on the surface of the NK cells. Such receptors include the CD94/NKG2 heterodimer receptor, Ly49, KIR (kill-cell immunoglobulin-like receptor), and ILT (leukocyte inhibitory receptor). Once CTLs and NKs recognize their target cells, they form with them a cell contact region reminiscent of a neural synapse, referred to as “immunological synapse,” and empty into this intercellular gap their secretory lysosomes (or lytic granules) (6, 7) (Figure 1A). The liberated granule hydrolases (i.e., granzymes) then enter the cytoplasm of the target cells with the assistance of another lysosomal protein, perforin, where they activate both caspase-dependent and -independent cell death pathways, finally causing their demise (4) (Figure 1B).
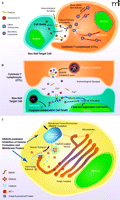
Formation and secretion of secretory lysosomes in the cytotoxic T-lymphocyte (CTL). A. Release of secretory lysosomes by CTLs when these cells encounter, recognize and interact with non-self cells. CTLs recognize non-self cells through foreign antigens co-presented with NHC class I molecules in the surface of target cells, forming with them an “immunological synapse.” Intra-CTL secretory lysosomes associate with microtubules and translocate towards the microtubule-organizing center (MTOC), accumulating at the vicinity of immunological synapses. Accumulated secretory lysosomes then fuse to the CTL plasma membrane and secrete their contents into the immunological synapses through the SNARE complex. Released perforin molecules allow entry of granzymes into the cytoplasm of the target cells, activating apoptotic pathways and leading to their demise. B. Secretory lysosome-mediated cell death by CTLs. Upon activation of CTLs, and after formation of the immunological synapse with non-self target cells, CTLs release the contents of secretory lysosomes (perforins and granzymes) into the lumen of the immunological synapse through reverse pinocytosis organized by the SNARE complex. Perforin then forms a complement-like transmembrane pore in the plasma membrane of target cells under the influence of Ca2+, allowing transfer of granzymes into these cells, in addition to uptake of granzymes by pinocytosis. Intracellularly accumulated granzymes stimulate cas-pase-dependent and -independent pathways to finally kill target cells (4). C. Formation and maturation of secretory lysosomes in CTLs. Lysosomal proteins, such as perforin and granzymes, are produced in the endoplasmic reticulum and transferred to the Golgi complex. These molecules then accumulate in the trans-Golgi network (TGN), where they interact with sorting molecules, the mannose 6-phosphatase receptors (M6PRs). TGN membranes containing lysosomal proteins eventually form clathrin-coated vesicles by attracting clathrins and adaptor proteins (APs) (e.g. AP-1). Clathrin-coated vesicles move along microtubules and fuse to maturing secretory lysosomes.
To better understand the ramifications of the Ruder et al. paper, some background discussion of the cell secretory process is necessary. Secretory lysosomes are released from both CTLs and NKs through the concerted operation of several intracellular secretory machineries (6, 7) (Figure 1A). Once these cells contact their target non-self cells, secretory lysosomes move along microtubules through interaction with kinesin- and dynein-motors and accumulate at the microtubule-organizing center (MTOC), which further migrates towards the immunological synapse. Secretory lysosomes subsequently fuse with the plasma membrane of the immunological synapse, under the regulation of the soluble N-ethylmaleimide-sensitive fusion factor attachment protein receptor (SNARE) complex. This protein complex, consisting of several molecules with extended α-helical structures (i.e., the syntaxins, synaptobrevins, and SNAP-25) residing in either vesicle or plasma membrane, functions by bringing the two membranes gradually into close apposition and eventually merging them like a “zipper” (6–8).
Production of mature secretory lysosomes in CTLs and NKs is achieved through a coordinated transfer of secretory molecules from the endoplasmic reticulum/Golgi complex into these vesicles (6, 9) (Figure 1C). The lysosomal molecules, including the perforins and the acid hydrolases granzymes, are produced in the endoplasmic reticulum and are subsequently transported to the Golgi complex (9). After these molecules undergo maturation inside the Golgi complex, they accumulate as “cargo” in the trans-Golgi-network (TGN), which is a tubular network-like organelle that emanates from the last trans-Golgi cistern, that appears to “peel off” from the rest of the Golgi stack (9, 10). At this particular portion of the Golgi complex, these cargo proteins bind to sorting proteins—the mannose 6-phosphate receptors (M6PRs)—through physical interaction between the latter’s extracytoplasmic domain and the M6P residues attached on the cargo molecules (10). As cargo-bound M6PRs pass through the TGN lipid bilayer membrane they may interact with adaptor proteins (APs) (10). The latter are heterotetramers consisting of two large subunits: a β subunit, a more divergent subunit (either α, γ, δ, or ɛ), a medium size μ subunit, and a small σ unit (11). Depending on the subunits employed, APs are divided into four subtypes (AP-1 to -4), each of which has specific activities in the transfer of proteins between different vesicles/cell compartments (12). For example, AP-1, which contains a γ subunit along with β1, μ1, and σ1, contributes to the formation of secretory lysosomes by facilitating transfer of lysosomal proteins from TGN to these vesicles (11). AP-1, in cooperation with a structurally similar AP-like molecule, termed the Golgi-localizing γ-adaptin ear homology domain Arf-binding protein (GGA), together with small GTP-binding proteins of the Arf, Rac1, and/or Rab families, binds to and attracts clathrin to form clathrin-coated vesicles that function as cargo carriers to the secretory lysosome and other subcellular compartments (9, 13). Clathrin-coated vesicles move along microtubules through interaction between adaptor proteins and kinesin-motors and deliver their contents into secretory lysosomes by fusing into the latter (9, 14). Because the protein transfer from TGN to secretory lysosomes is a key component for production and maturation of these vesicles, this process is highly regulated by several distinct cell pathways through modification of its components, such as by phosphorylation, glycosylation, and ubiquitination, as well as by alteration of the lipid composition in the TGN membrane (9).
Rüder et al. reported that the estrogen receptor-binding fragment-associated gene 9 (EBAG9) inhibits the CTL-mediated adaptive immune response by negatively regulating the cytolytic activity of CTLs (1) (Figure 1C). EBAG9, also called receptor binding cancer antigen expressed in SiSO cells (RCAS1), was originally identified as an estrogen-inducible gene in the screening of the MCF7 CpG-island library (15). Exploration of gene promoters that physically interacted with the DNA-binding domain of the estrogen receptor identified EBAG9, while subsequent analyses revealed a classic, tandem estrogen response element in the promoter region of EBAG9 (15). Human EBAG9 consists of 213 amino-acids and is expressed in most tissues and cells (2). It forms homooligomers through its C-terminal coiled-coil structures and is exclusively accumulated in the Golgi complex (2). It is noteworthy that tissue EBAG9 expression is increased in various malignancies, including cancers of the gastro-intestinal tract, hepatocellular carcinoma, non-small cell lung cancers, and breast, ovarian, endometrial, and cervical cancers, while its tissue expression is negatively correlated with the prognosis of patients bearing these malignancies (2). EBAG9 is also a secretory protein detected in the sera of cancer patients (2). In their manuscript, Rüder et al. demonstrated that loss of EBAG9 expression enhanced the cytolytic activity of CTLs in vivo by increasing the release of secretory lysosomes from these cells (1). EBAG9 physically interacted with the γ2 subunit of AP-1 and negatively regulated transfer of lysosomal proteins from the TGN to secretory lysosomes, resulting in formation of smaller secretory granules, whereas it did not affect the translocation of these vesicles along microtubules to MTOC or the formation of the immunological synapse upon stimulation of CTLs (1). These results indicated that EBAG9 is a negative regulator of vesicle transfer from TGN to secretory lysosomes, most likely by inhibiting the activity of AP-1 on clathrin-coated cargo formation/transfer. Indeed, EBAG9 is the first molecule ever reported to negatively regulate protein transfer from TGN to secretory lysosomes (1, 6).
Each tissue employs specific molecules and/or their isoforms for transferring and secreting different vesicles (9). In agreement with this observation, in an earlier study, Rüder et al. demonstrated that EBAG9 negatively regulated exocytosis of neuropeptide Y in PC12 neuroblastoma cells, but not in mouse primary hippocampal neurons, suggesting tissue or cell type specificity in its actions (16). They further found, using a yeast two-hybrid screening assay, that EBAG9 physically interacted with Snapin, which is a modulator of the SNARE complex that governs exocytosis in neuronal cells (16) (Figure 1C). Although the authors did not examine the importance of the physical interaction of EBAG9 with AP-1 and Snapin in their respective biologic functions and provided no mechanistic explanation for their molecular interaction (1, 16), it appears that EBAG9 is a negative regulator of the secretion of intracellular vesicles by suppressing cargo transfer, as well as fusion/exocytosis of these vesicles, by targeting their partner molecules.
Various cancers, including those found in estrogen-responsive organs and tissues, have elevated expression of EBAG9 inside tumor cells (2). Previous reports indicated that EBAG9 produced in cancer cells was secreted into the interstitial space and functioned as a ligand for a putative receptor present on numerous cells, including T-, B-lymphocytes and NKs, inducing their apoptosis and death, which enabled cancer cells to evade immune surveillance (2, 17, 18). Secreted EBAG9 also enhanced invasive potency of cancer cells by induction of stromal tissue remodeling and by changing surface expression of tumor associated O-linked glycans, which could participate in cell adhesion, invasion and metastasis of these cells (19, 20). However, the majority of EBAG9 is expressed inside tumor cells, and as a result, a question comes up as to how such intracellular EBAG9 expressed in tumor cells plays a role in their biology in association with its newly identified activity on vesicle formation and secretion. First, EBAG9 might modulate cell surface expression of the cation-independent (CI) form of M6PR, by suppressing formation and/or exocytosis of the vesicles containing this receptor through targeting AP-1. Overexpression of CI-M6PR was shown to suppress cellular growth in vitro and in vivo (21, 22), while cell surface CI-M6PR regulates cell growth by changing the clearance of insulin-like growth factor II and by facilitating maturation of transforming growth factor (TGF) β (23). Further, increased production of EBAG9 might alter the expression of MHC class I molecules on the surface of tumor cells: MHC class I molecules are produced in the endoplasmic reticulum, modified in the Golgi complex and transferred to the plasma membrane as a component of recycling endosomes (24). MHC class I abnormalities, such as structural alterations, including total, haplotype, and allelic loss of the MHC class I heavy chain and dysregulation of various components of the MHC class I antigen processing machinery, have been found both in solid and hematologic tumors of various tissue origins (24). Such alterations in MHC class I expression in tumor cells may in part be caused by elevated levels of intracellular EBAG9, presumably providing an advantage to tumor cells to escape from cytolysis promoted by CTLs and/or NKs. EBAG9 could be particularly important for the development of malignancies under estrogen regulation, such as those originating from breast endothelial cells, as estrogens increase EBAG9 expression, as well as support growth of malignant cells and increase the incidence of such malignancies (25). EBAG9 expressed in response to elevated levels of estrogens in cytotoxic CTLs and NKs would further provide an advantage to the development of these malignancies, as elevated EBAG9 in these immune cells suppresses their cytotoxic activity by reducing formation/exocytosis of secretory lysosomes. Indeed, over ten years ago, anti-estrogens, such as tamoxifen and toremifene, were reported to augment immune oncolysis by NKs and CTLs, whereas a more recent study indicated that EBAG9 promoted growth and distant metastases of 4T1 mouse mammary carcinoma cells by decreasing CD8+ T cell infiltration into the tumor and their responsiveness against tumor cells (26–28). Thus, anti-estrogens not only inhibit tumor growth directly, but may also employ EBAG9 as a molecular switch to activate NKs and CTLs against the tumor. Further research will shed more light on the pathophysiologic importance of EBGA9 in the development, growth and spread of malignancies, in strong connection with its involvement in intracellular vesicle transport and cytotoxic immunity.
Acknowledgments
This work was supported by the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD, and the University of Athens, Athens Greece.
- Copyright © 2009
References

Tomoshige Kino, MD, PhD, is Head, Unit of Molecular Hormone Action, Program in Reproductive and Adult Endocrinology, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health. He received his Doctorate thesis at the Chiba University Medical School, Japan, and completed clinical training at the same University. He then joined to Dr. Chrousos’s laboratory at the NICHD, NIH. His research focuses on the understanding of the mechanisms of action of nuclear hormones, the influences of these hormones on metabolic, immune and neuronal functions and their involvement in human physiology and disease. He has published more than 100 articles.

George P. Chrousos, MD, is Professor and Chairman of the First Department of Pediatrics, University of Athens Medical School, Greece, and Scientist Emeritus at the National Institutes of Health, Bethesda, Maryland. He completed his Doctorate thesis at the University of Athens and did his residency in Pediatrics at New York University Medical School, New York, NY, and his fellowship in Endocrinology at the Clinical Center of the National Institutes of Health, Bethesda, Maryland. Before his return to Greece, Dr. Chrousos was consecutively Senior Investigator, Head of the Pediatric Endocrinology Section and Training Program, and Chief of the Pediatric and Reproductive Endocrinology Branch of the NICHD. During his time at the NIH, Dr. Chrousos also held the position of Clinical Professor of Pediatrics, Physiology and Biophysics at Georgetown University Medical School. His work has led to improved understanding of the physiology and pathophysiology of the stress response at the neuroendocrine, cellular, and molecular levels. He has also advanced the study of hypothalamic-pituitary-adrenal axis diseases and has made major contributions to the understanding of nuclear receptor signaling systems. He has authored and coauthored more than 1,000 scientific publications, has edited twenty-five books and his work has been cited more than 47,000 times. He is currently the President of the European Society of Clinical Investigation and the Greek College of Pediatrics. E-mail chrousge{at}med.uoa.gr; fax +30-210-7794023.

