Wishing Away Inflammation? New Links between Serotonin and TNF Signaling
The idea that emotional states can influence health has been widely accepted in medicine since Roman times (1). This idea has been validated in the modern era; several neuroendocrine pathways have been elucidated that link neural and immune responses (2). These links function reciprocally: a number of neural signaling pathways trigger the release of immunomodulators, and cells of the nervous system sense inflammation through receptors for immune cytokines. The Hypothalamic-Pituitary-Adrenal Axis “translates” psychological stressors into glucocorticoid synthesis by the adrenal cortex and epinephrine synthesis by the adrenal medulla, leading to an immediate epinephrine-activated vasoactive response. Glucocorticoid-mediated immune modulation may provide the short-term advantage of suppressing inflammation during stress, whereas chronic endogenous production or exogenous administration of glucocorticoids results in immunosuppression. Both the benefits and detriments of these effects are seen in patients placed on glucocorticoid therapy for autoimmune and inflammatory diseases. A recent paper by Yu et al. identifies a novel pathway of crosstalk between neural and immune receptors with the observation that pharmacological agonists of 5-hydroxytryptamine2A (5-HT2A) receptors can block the pro-inflammatory effects of Tumor Necrosis Factor (TNF) on smooth muscle vascular cells (3).
5-Hydroxytryptamine, commonly known as serotonin, is a neurotransmitter with potent effects on many aspects of mood and cognition. Selective serotonin reuptake inhibitors [(SSRIs), see (4)] are thought to mediate their antidepressant effects through enhancing the availability of serotonin in the CNS. Of the seven subtypes of serotonin receptors identified in humans, only the 5-HT2A receptor is involved in cognition. Hallucinogens such as lysergic acid diethylamide (LSD) are thought to exert their actions through 5-HT2A receptors, and atypical antipsychotics and some newer generation anti-depressants are thought to act primarily as 5-HT2A antagonists. For these reasons, the effects of serotonin on the CNS have been intensively studied. However, most serotonin is actually produced in other parts of the body by various cells types such as activated intestinal enterochromaffin cells, mast cells, and platelets. Serotonin regulates numerous biological processes outside the CNS such as bowel motility, platelet aggregation, cardiac function, and bladder control (5). In addition, serotonin has important effects on vascular and uterine smooth muscles, as it is involved in their growth and contraction (6). In the immune system, serotonin activates human monocytes and prevents their apoptosis (7) and modulates cytokine and chemokine production in lipopolysaccharide (LPS)-primed monocytes (8). In vivo, serotonin appears to be pro-inflammatory, as a number of studies have shown depletion of serotonin within the CNS acts to reduce animal models of inflammation such as adjuvant-induced arthritis (9–11).
In contrast to the apparently pro-inflammatory role of serotonin as a neurotransmitter, Yu et al. (3) show that activation of 5-HT2A receptors on smooth muscle cells with the synthetic agonist (R)-1-(2,5-dimethoxy-4-iodophenyl)-2-aminopropane [(R)-DOI] suppresses multiple responses to TNF. (R)-DOI repressed the TNF-mediated induction of adhesion molecules and cytokine production at concentrations in the low picomolar range, which might be considered “superpotent” because this is below the nanomolar affinity measured for (R)-DOI at 5-HT2A receptors. (R)-DOI suppressed TNFα-mediated induction of mRNA for proinflammatory markers, such as VCAM-1, ICAM-1, and IL-6, nitric oxide synthase activity, and translocation of NF-κB all at this very low concentration. These effects were restricted to 5-HT2A receptors, as 5-HT2B and 5-HT2C receptor-selective agonists were ineffective in suppressing TNFα-induced inflammation. (R)-DOI was also compared with three other 5-HT2A receptor agonists: the phenethylamine 2C-BCB [(4-bromo-3,6-dimethoxybenzocyclobuten-1-yl) methylamine]; and two indolealkylamines: LA-SS-Az [(2′S,4′S)-(+)-9,10-didehydro-6-methylergoline-8β-(trans-2,4-dimethylazetidide)] and LSD. These three agents suppressed TNF-induced ICAM-1, V-CAM-1, and IL-6 expression, but with less potency than (R)-DOI. In addition, (R)-DOI was able to significantly inhibit the effects of TNFα when administered up to four hours after TNFα, indicating possibly therapeutic effects of (R)-DOI on already established chronic inflammation.
Yu et al. (3) only hint at the signal transduction pathways underlying blockade of TNF signaling by the 5-HT2A agonists. TNF has two receptors, TNFR1 (p55) and TNFR2 (p75), but most of the pro-inflammatory effects of TNF are mediated through TNFR1, suggesting that 5-HT2A agonists likely act on TNFR1. Inhibitor studies suggested that the effects of 5-HT2A agonists are mediated through the activity of protein kinase C family members. TNFR1 signaling was affected by inhibition of NF-κB translocation to the nucleus, suggesting a direct effect of (R)-DOI on early events in TNFR1 signaling. The primary TNFR1 signaling complex consists of the adapter protein TNFR1-associated death domain protein (TRADD), the ring finger–containing proteins TNF receptor-associated factor 1 (TRAF1) and TRAF2, and the protein kinase receptor-interacting serine–threonine protein kinase (RIP), which act together to activate the IκB Kinase complex to trigger degradation of the inhibitor of NF-κB (IκB) and release of NF-κB subunits into the nucleus where they function as transcription factors (Figure 1). Interestingly, protein kinase C (PKC) agonists, such as Phorbol 12-Myristate 13-Acetate (PMA, also termed TPA) can block TNF signaling and inhibit assembly of the TNFR1 proximal signaling complex (12).
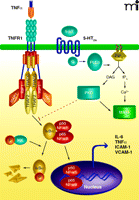
Potential mechanism of crosstalk between the tumor necrosis factor receptor 1 (TNFR1) and serotonin subtype 2A (5-HT2A) receptor signaling. The primary TNFR1 signaling complex consists of the adapter protein TRADD, the ring finger–containing protein TRAF2, the inhibitor of apoptosis protein cIAP1, and the protein kinase RIP1. Following TNF binding, the receptor complex activates the IκB kinase complex to trigger IκB degradation and the release of NF-κB subunits into the nucleus where they function as transcription factors. The TNF-bound receptor complex also induces the expression of several pro-inflammatory genes such as IL-6, TNFα, ICAM-1, and VCAM-1. The receptor for serotonin, 5-HT2A is a G protein–coupled receptor able to stimulate phospholipase C (PLC), a membrane-bound enzyme that catalyzes the degradation of the inositol lipid phosphatidylinositol 4,5-bisphosphate (PIP2), producing inositol 1,4,5-triphosphate (IP3) and diacylglycerol (DAG). IP3 mobilizes Ca2+ that induces multiple responses in the cell, including activation of mitogen-activated protein kinase (MAPK), while DAG activates another kinase family, protein kinase C (PKC). Results from Yu and colleagues (3) suggest that 5-HT2A receptor-mediated anti-inflammatory effects are mediated through activation of PKC, which acts most probably at a level proximal to NF-κB nuclear translocation. TRADD, TNFR1-associated death domain protein; TRAF2, TNF receptor-associated factor 2; cIAP1, cellular inhibitor of apotosis 1; RIP, receptor-interacting serine–threonine protein kinase; IL-6, interleukin-6; ICAM-1, intercellular adhesion molecule-1; VCAM- 1, vascular cell adhesion molecule-1.
Although these results suggest that 5-HT2A agonists might be investigated further as an anti-inflammatory agent, there are a number of concerns that limit interpretation of these results. The suppressive effects of (R)-DOI on induction of pro-inflammatory genes were only reported for mRNAs. It is certainly necessary to determine whether these effects operate at the protein level as well. Most experiments were done with (R)-DOI, or other synthetic 5-HT2A receptor agonists. It would be useful to know whether these effects are seen with serotonin itself and whether they could be reversed by a 5-HT2A receptor antagonist such as ketanserin or by using cells deficient in the 5-HT2A receptor. Additionally, it is essential to test whether the observed effects of (R)-DOI on TNFα-induced inflammation also apply to other cell types, especially primary human cells responsive to TNF such as hepatocytes, endothelial cells, and monocytes.
Despite these concerns, it is tempting to speculate on the clinical implications of this study. Single-nucleotide genetic polymorphisms in the 5-HT2A receptor gene have been associated with rheumatoid arthritis (13), although the genome-wide significance of this association is not clear. Although not tested in this study, one might predict from these results that patients on 5-HT2A-blocking agents would become hypersensitive to the effects of TNF and possibly develop arthritic symptoms. In fact, in a recent retrospective survey of drug reactions, Dahlqvist and colleagues found a 45-fold excess rate of joint complaints in patients given 5-HT2A-blocking antidepressants, such as mianserin, nefazodone, and mirtazapin compared to patients given SSRIs (14). The same group observed that the density of serotonin 5-HT2A receptors was decreased in rheumatoid arthritis patients (15), suggesting a possible role for reduced expression of the 5-HT2A receptor in the sensitization of cells to TNF in rheumatoid arthritis. One might also speculate that SSRIs could be anti-inflammatory, although there is little evidence of this in the literature, and SSRIs would increase availability of serotonin for all subgroups of receptors, not just 5-HT2A.
Stimulating 5-HT2A receptors to suppress inflammation would clearly require the development of agents that do not affect the CNS, as 5-HT2A agonists such as LSD are well known to induce hallucinations and altered mental status. Also, the discovery that (R)-DOI was able to potently inhibit the effects of TNFα even for up to two hours after the administration of TNFα indicate that potential therapies could not only be aimed at preventing inflammation but also treating inflammatory injury that has already occurred or is ongoing. However, it would be important to determine the duration of the effects and whether receptor desensitization occurs, because treatment with 5-HT2A agonists would most likely be needed for much longer periods of time in patients with chronic inflammatory joint disease. Whether these results bear fruit in terms of therapeutic agents, they add one more thread to the intriguing web of interconnections that link our immune and nervous systems.
Acknowledgments
Funded by NIAMS intramural research program. We would like to thank Ariel C. Bulua for critical reading of this article.
- Copyright © 2009
References

Richard Siegel’s interest in immunology, autoimmunity, and apoptosis began as an MD/PhD student at the University of Pennsylvania School of Medicine, from which he graduated in 1993. He trained in Internal Medicine and Rheumatology at Hospital of the University of Pennsylvania, and then moved to the NIH to do postdoctoral training 1996, where he worked in Michael Lenardo’s laboratory in NIAID studying the molecular basis of autoimmunity in the Autoimmune Lymphoproliferative Syndrome (ALPS). In 2001, Dr. Siegel moved to the National Institute of Arthritis, Musculoskeletal Diseases and Skin (NIAMS) at the NIH as an Investigator, where he also attends on the Rheumatology service at the NIH Clinical Center. He is presently Senior Investigator and Acting Chief of the Autoimmunity Branch in NIAMS, where he directs the Immunoregulation Section. The Siegel lab seeks to understand how alterations in regulatory signaling pathways in immune cells lead to abnormal immune responses, chronic inflammation, and autoimmune diseases. The lab has focused principally on the biology of TNF-family cytokines in normal and pathological immune responses. More about the lab can be found at www.niams.nih.gov/Research/Ongoing_Research/Branch_Lab/Autoimmunity/irg.asp. E-mail siegelr{at}mail.nih.gov; fax 301-451-5394.

Martin Pelletier, PhD, received his doctorate in 2005 in Virology and Immunology at INRS-Institut Armand-Frappier (Laval, Quebec, Canada) for his research in cytokine-mediated neutrophil activation by interleukin-15 and interleukin-21. As a postdoctoral fellow at Università Degli Studi di Verona (Verona, Italy), he evaluated the cross-talk between human neutrophils and Th17 cells. He is currently a Research Fellow in Richard Siegel’s laboratory at National Institute of Arthritis and Musculoskeletal and Skin Diseases, NIH (Bethesda, Maryland, USA) studying the inflammatory processes involved in the autoinflammatory disorder called tumor necrosis factor receptor-associated periodic syndrome (TRAPS). E-mail: martin.pelletier{at}nih.gov

