|
 
 |
|
CASE REPORT |
|
|
|
| Year : 2011 | Volume
: 17
| Issue : 1 | Page : 26-28 |
| |
Familial clustering of a rare syndrome
Jayashree Nadkarni, Hari Ganesh, Rashmi Dwivedi
Department of Pediatrics, Gandhi Medical College and Associated Kamla Nehru Hospital, Bhopal - 462 016, M.P, India
| Date of Web Publication | 18-Jun-2011 |
Correspondence Address:
Jayashree Nadkarni
3rd Floor, Kamla Nehru Hospital, Bhopal - 462 016, M.P
India
 Source of Support: None, Conflict of Interest: None
DOI: 10.4103/0971-6866.82189

 Abstract Abstract | | |
Ectrodactyly, ectodermal dysplasia and cleft palate syndrome is a rare autosomal dominant multiple congenital anomaly syndrome with variable expressivity and reduced penetration. The cardinal features are cleft palate/lip, lobster hand deformity, sparse hypopigmented hair, dry scaly skin, and lacrimal and urogenital anomalies. A neonate presented to us with typical features, his mother and other two siblings were also affected.
Keywords: EEC, ectrodactyly, variable presentations
How to cite this article:
Nadkarni J, Ganesh H, Dwivedi R. Familial clustering of a rare syndrome. Indian J Hum Genet 2011;17:26-8 |
| Introduction | |  |
The term EEC syndrome was coined by Rudiger et al., and consists of ectrodactyly (E) and ectodermal dysplasia (E), and cleft lip/palate (C). [1] Other features include peg-shaped teeth with enamel hypoplasia, conductive deafness, lacrimal duct, and urogenital and neurological anomalies. The syndrome results from a developmental abnormality that simultaneously affects the ectodermal and mesodermal tissues. [2] Reports of the classical triad of anomalies in families, especially in Indian literature, are few. The inheritance of this condition is autosomal dominant, but there is genetic heterogeneity, variable expressivity, and reduced penetrance accounting for the clinical variability even in the same families, thus making genetic counseling difficult.
| Case Report | | |
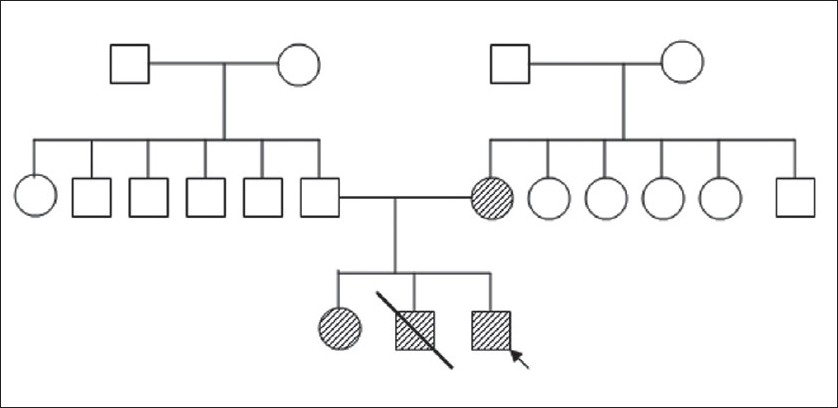
A one-day-old neonate was referred to our centre for multiple congenital anomalies and prematurity. He was the third among the offsprings, born to nonconsanguineous parents. At the time of conception, mother's and father's ages were 26 and 34 years, respectively. It was a 35 week preterm neonate born by normal vaginal hospital delivery with no significant antenatal history. At birth, the baby had immediate cry.
On examination, he weighed 1.6 kg, with a length of 41 cm and head circumference was 30.5 cm. The skin was dry and scaly, more so on the upper trunk, face, hands, and feet. The hair was sparse, lusterless, and hypopigmented with frontal alopecia. Eyebrows were also thin and sparse. Skeletal anomalies were ectrodactyly with a typical lobster/claw hand--foot deformity. He also had unilateral cleft lip and palate. There was no midfacial hypo- or hyperplasia. Ophthalmological evaluation revealed loss of cilia, with a wrinkled appearance of upper and lower lids on both sides and normal lacrimal puncta. The visual acuity, cornea, fundus, and inner and outer canthal distances were normal. There were no auditory, urogenital, or neurological anomalies on clinical examination.
Routine investigations were within normal limits. X-rays of the hands and feet showed radiological findings corresponding to the clinical features. X-ray spine ruled out spina bifida. There was no urogenital anomaly on ultrasonography.
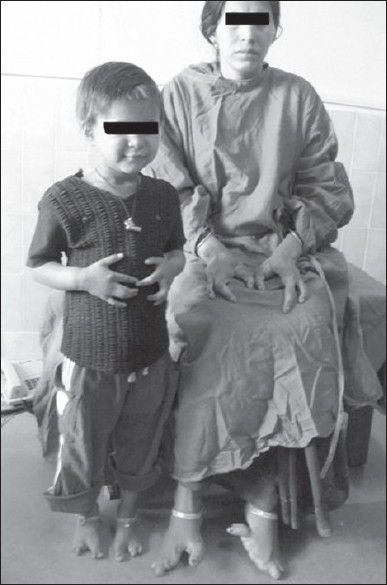
Similar skeletal abnormalities were present in the mother and the elder sibling (5-year-old female), though they did not have cleft palate [Figure 1] The second sibling died at the age of 4 days and according to the mother, he also had similar hand deformities, but no orofacial deformity [Figure 2].
| Discussion | | |
The EEC syndrome is a rare autosomal dominant disorder with variable expressivity and reduced penetrance. [3] The central reduction defect of the hands and feet known as ectrodactyly, split hand/split foot malformation, or lobster-claw, occurs in approximately 1 in 18 000 newborns. Other modes of inheritance have also been noted. The karyotype is usually normal. This disorder has been attributed to mutations in a gene encoding the p63. P63 is a transcription factor that regulates the activity of the tumor suppressor gene TP53. Based on linkage studies and the analysis of chromosomal abnormalities, three loci have been identified in humans. [4] It is a type of ectodermal dysplasia with variable involvement of skin, hair, teeth, and nails [Figure 3]. Ectodermal dysplasia may manifest as diffuse dryness and hypopigmentation of skin, hypopigmented and scanty hair, dysplastic nails, and abnormal dentition. Ophthalmologic manifestations include entropion, telecanthus, absence of lacrimal puncta, absence of lashes, blepharitis, corneal opacification, and neovascularization. Other ectodermal anomalies include mild hypohidrosis; coarse, dry hair with hypotrichosis; xerostomia; dystrophic nails; dental enamel hypoplasia; and microdontia. [5] Associated defects include conductive deafness, choanal atresia, and abnormalities of the genitourinary tract such as megaureter, duplicating collecting system, diverticuli, ureterocele, etc. [6]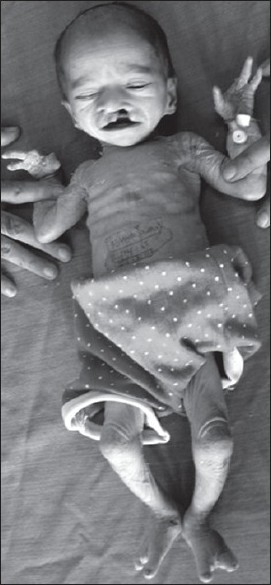 | Figure 3: Showing dry scaly skin, sparse hair and eyebrows, cleft palate and limb deformities
Click here to view |
Ectrodactyly refers to variable deformities of hand and feet, having a common central split or cleft, but its occurrence is not obligatory. In a meta-analysis of 230 published cases (116 familial and 114 sporadic), ectrodactyly was found in 84%, ectodermal dysplasia in 77%, clefts in 68%, and anomalies of lacrimal ducts in 59%. Urogenital defects were reported in 52 patients and conductive hearing loss in 33. Isolated cases were more severely affected than familial cases. [7] Our case presented with all the three cardinal features but no eye or genitourinary abnormalities. It is probable that some of the manifestations may take some time to develop, hence are not seen in this neonate. Also, variability in the phenotype within the same family is seen. Anneren et al., suggested that low birth weight and polysyndactyly (without ectrodactyly) may be features of the EEC syndrome. [8] Jindal G. reported a 6 and half-year-old boy with similar hand foot deformity whose sibling, a term female stillbirth also had a similar anomaly, but neither had other associated defects. [9]
Fryns et al., reported variable manifestations of EEC in 2 families, confirming that no symptom is obligatory for the diagnosis. In the absence of cleft lip/palate, EEC patients have a characteristic facial morphology with maxillary hypoplasia, short philtrum, and broad nasal tip. Interfamilial variability was significantly greater than intrafamilial variability, pointing to genetic (allelic?) heterogeneity. The penetrance of the EEC mutation is between 93 and 98%. No signs of anticipation were apparent. [10] The variable expressivity accounts for phenotypic variability in the affected members of the same family. Management of the cases requires multidisciplinary approach and reassurance regarding the low risk of mental handicap.
| References | | |
| 1. | Rudiger RA, Haase W, Passrge E. Association of ectrodactyly, ectodermal dysplasia and cleft lip/palate. Am J Dis Child 1970;120:160-3. 
|
| 2. | Smith S. Recognizable patterns of human malformations, 6 th ed. USA: Elsevier Saunders Publication; 2006. p. 330.
|
| 3. | Walker JC, Clodius L. The syndromes of cleft lip, cleft palate and lobster claw deformities of hands and feet. Plast Reconst Surg 1963;32:627-36.
[PUBMED] |
| 4. | Celli J, Duijf P, Hamel BC, Bamshad M, Kramer B, Smits AP, et al. Heterozygous germline mutations in the p53 homolog p63 are the cause of EEC syndrome. Cell 1999;99:143-53.
[PUBMED] [FULLTEXT] |
| 5. | Buss PW, Hughes HE, Clarke A. Twenty-four cases of the EEC syndrome: Clinical presentation and management. J Med Genet 1995;32:716-23.
[PUBMED] [FULLTEXT] |
| 6. | Nardi AC, Ferreira U, Netto NR Jr, Magna LA, Rodini ES, Richieri-Costa A. Urinary tract involvement in EEC syndrome: A clinical study in 25 Brazillian patients. Am J Med Genet 1992;44:803-6.
|
| 7. | Roelfsema NM, Cobben JM. The EEC syndrome: A literature study. Clin Dysmorph 1996;5:115-27.
[PUBMED] |
| 8. | Anneren G, Andersson T, Lindgren PG, Kjartansson S. Ectrodactyly, ectodermal dysplasia and cleft lip/palate (EEC): The clinical variation and prenatal diagnosis. Clin Genet 1991;40:257-62.
|
| 9. | Jindal G, Parmar VR, Gupta VK. Ectrodactyly/split hand feet malformation. Indian J Hum Genet 2009;15:140-2.
[PUBMED]  |
| 10. | Fryns JP, Legius E, Dereymaeker AM, Van den Berghe H. EEC syndrome without ectrodactyly: Report of two new families. J Med Genet 1990;27:165-8.
[PUBMED] [FULLTEXT] |
[Figure 1], [Figure 2], [Figure 3]
|